![]()
|
ACTUALIZACIÓN DE LAS DEMENCIAS FRONTOTEMPORALES Descargar en pdf. Ver diapositivas Power Point Autores: Mª Elena Toribio Díaz1, Jaume Morera Guitart2. 1 Unidad de Neurología de la Conducta y Demencias (CDP-Alz). Hospital San Vicente. San Vicente del Raspeig. Alicante. 2 Hospital La Pedrera. Denia. Alicante. Correspondencia: Mª Elena Toribio Díaz. Hospital San Vicente. C/ Lillo Juan, 137. San Vicente del Raspeig. Alicante. E-mail: etoribiod@hotmail.com Introducción En los últimos años se ha producido un notable avance en el conocimiento de las Demencias Lobares Frontotemporales (DLFT). El objetivo que nos proponemos con este trabajo es la realización de una revisión de los avances más recientes, descritos en la literatura científica, condensando de una manera inteligible toda la información sobre los aspectos clínicos, genéticos e histológicos de este grupo de enfermedades. Con el término Demencia Lobar Frontotemporal (DLFT) se hace referencia a un grupo de enfermedades neurodegenerativas muy heterogéneo tanto por su presentación clínica, por su componente genético y sus características histopatológicas. Constituye la tercera causa de demencia degenerativa después de la Enfermedad de Alzheimer (EA) y la Demencia con cuerpos de Lewy, la segunda en personas menores de 65 años. Su debut suele situarse entre los 45 y los 65 años con una distribución similar entre ambos sexos. Se describe una historia familiar positiva entre un 30 y un 45% de los casos [[1]]. Tres son los rasgos clínicos que la caracterizan: alteración del comportamiento con cambio en la personalidad, alteración de la conducta social y afectación del lenguaje [[2]]. En ocasiones algunos pacientes pueden presentar un cuadro de parkinsonismo o de enfermedad de motoneurona asociados [[3]]. La perspectiva histórica de las DLFT ha venido caracterizada por etapas de gran interés por definirla como una entidad independiente, frente a otras en las que era considerada una variante de la Enfermdad de Alzheimer (EA). La primera mención a este tipo de demencia se debe a Arnold Pick, quien en 1892 hizo la descripción de un varón de 71 años con un cuadro progresivo de deterioro mental al que se asociaba una afasia grave, en cuya necropsia se pudo constatar una marcada atrofia cortical del lóbulo temporal izquierdo. Por primera vez se planteaba la posibilidad de un síndrome demencial originado por una atrofia cerebral focal, y no por una atrofia generalizada de acuerdo a las tendencias de la época. En los años siguientes continuaron las descripciones histológicas y clínicas por parte de diferentes autores (Pick, Spielmeyer, Scheneider o el propio Alzheimer, entre muchos otros), si bien acabó perdiéndose el interés por este tipo de demencia en las primeras décadas del siglo XX, al ser considerada como una variante de la EA [[4]] En los años 80 renace de nuevo el interés por esta entidad: Lund, Brun y Gustafson (1986) la denominan Degeneración Lobar Frontotemporal [[5],[6]]. Neary y colaboradores (1988) Demencia de tipo frontal [[7]]. Paulatinamente se fueron añadiendo nuevas entidades clínico-patológicas a este heterogéneo grupo de demencias tipo no Alzheimer como la Degeneración Corticobasal, la DLFT con parkinsonismo o la DLFT asociada a enfermedad de motoneurona. En 1982 Mesulam [[8]] describió una serie de pacientes con un trastorno del lenguaje en forma de afasia lentamente progresiva sin afectación cognitiva o conductual y al que denominó Afasia Lentamente Progresiva Sin Demencia. Con la evolución de la enfermedad estos pacientes acabaron desarrollando una demencia de tipo frontal como se pudo comprobar posteriormente. En 1989 Snowden [[9]] describió la Demencia Semántica como un cuadro de afasia progresiva causado por un déficit selectivo en la memoria semántica completándose así los cuadros clínicos principales de la DLFT. Es, sin embargo, en los últimos 20 años donde encontramos el mayor avance en la investigación en este tipo de demencia: por una parte los avances genéticos con el descubrimiento del gen MAPT (1998) [[10]] y el gen de la Progranulina (2006) [[11],[12]] así como de sus mutaciones responsables de un porcentaje elevado de las DLFT hereditarias. Por otra, el desarrollo de nuevas técnicas inmunohistoquímicas con la detección de la proteína TDP-43 (2006) [[13]] como principal integrante de las inclusiones neuronales en las DLFT tau negativas y finalmente el desarrollo de nuevas técnicas de neuroimagen como la RMN funcional, SPECT o PET [[14]]. La característica principal de la DLFT es su gran heterogeneidad lo que a nivel clínico se traduce en la existencia de varios síndromes con unos síntomas comunes, como son los cambios en la personalidad o en la conducta social del paciente asociados a una afectación del lenguaje. Se describen tres subtipos clínicos principales en función de la predominancia de cada uno de estos síntomas o de su momento de aparición a lo largo de la evolución de la enfermedad: Demencia frontotemporal variante frontal, Demencia Semántica y Afasia Primaria Progresiva No Fluente [2]. La Demencia Frontotemporal variante frontal (DFT) constituye el síndrome clínico más frecuente. Su característica principal es un cambio insidioso en la personalidad del paciente adoptando un comportamiento inusual como si fuera otra persona, o por el contrario acentuándose de manera ostensible ciertos rasgos de su carácter. Las alteraciones son variables y dependen de las áreas prefrontales afectas [[15]] conformando tres síndromes prefrontales o frontosubcorticales típicos: Dorsolateral, Orbitomedial y Mesiofrontal (tabla 1).
El cuadro clínico en la DFT se completa con una pérdida progresiva de la capacidad para la expresión del lenguaje con ecolalia, perseveración y alteración tanto de la nominación como de la escritura. De forma típica se produce una preservación de la memoria hasta etapas algo más avanzadas. En la Demencia Semántica se produce una desintegración de la base de conocimientos que sostienen el lenguaje, la memoria semántica, por lo que los pacientes son incapaces de reconocer los objetos, hechos, palabras o su significado. Dado que los aspectos fonológicos y sintácticos permanecen intactos se produce un lenguaje espontáneo fluente pero con escaso contenido informativo, un déficit tanto de la nominación como de la comprensión y parafasias semánticas [[17]]. En la Afasia Primaria Progresiva No Fluente hay una alteración de los aspectos fonológicos y sintácticos del lenguaje por lo que la alteración en la fluidez será muy marcada con parafasias fonéticas, comprensión conservada en fases iniciales, agramatismo y anomia que acaba por evolucionar al mutismo. Finalmente cabe reseñar un tercer subtipo de trastorno del lenguaje en las DLFT denominado por Gorno-Tempini [[18]] como Variante Logopénica caracterizada por una severa afectación de la nominación, la fluidez, la repetición y la comprensión sintáctica compleja, con un preservación característica de la comprensión de la palabra aislada con escasa presencia de parafasias. De forma típica el trastorno progresivo del lenguaje (diapositiva 1) constituye la única manifestación durante los primeros dos años con el desarrollo posterior de la afectación conductual [[19]] y de la memoria [2]. Dificultades para la definición de unos criterios diagnósticos definitivos La heterogeneidad clínica e histológica de los distintos síndromes englobados en la DLFT justifica la necesidad de definir unos criterios diagnósticos que faciliten su identificación, pero esta heterogeneidad supone en sí misma la mayor dificultad para lograrlo (diapositiva 2), por lo que son diversos los criterios propuestos por varios grupos de investigadores: 1. Los grupos de Lund y Manchester [[20]] elaboraron los primeros criterios clínico-patológicos cuya principal aportación fue una buena discriminación frente a la EA. Sin embargo no establecían el número de síntomas necesarios o la importancia relativa de cada uno de ellos en el diagnóstico como tampoco aportaban una definición operativa de los mismos. 2. Estas limitaciones llevaron al desarrollo de nuevos criterios por parte del grupo de Neary [2] con la definición de las características clínicas específicas de los tres síndromes principales de la DLFT: Demencia frontotemporal variante frontal (DFT), Afasia Primaria Progresiva No Fluente (APPNF) y Demencia Semántica (DS). La precisión diagnóstica antemortem de estos criterios se ha podido confirmar mostrando una sensibilidad del 85% y una especificidad del 99% [[21]]. 3. No obstante para algunos autores se hacía necesaria la simplificación de unos criterios poco funcionales para el clínico no especializado, por lo que Mackhann y colaboradores [[22]] agruparon en el concepto Demencia Frontotemporal a aquellos síndromes caracterizados por un cuadro precoz de cambio en la personalidad o de alteración del lenguaje, pero con el inconveniente de no poder discriminar aquellas formas de EA con afectación circunscrita de este último. 4. Finalmente en los últimos criterios diagnósticos propuestos por el grupo de Cairns [[23]] en el año 2007 se establece una nueva clasificación atendiendo a los recientes avances en genética molecular, bioquímica y neuropatología. Clasificación histopatológica y genética Tradicionalmente se ha considerado la DLFT como un conjunto de procesos neuropatológicos con degeneración predominante de los lóbulos frontales y temporales compartiendo un fenotipo clínico similar con algunas diferencias en función de la distribución topográfica del trastorno patológico subyacente [[24]]. A nivel histopatológico se ha podido constatar la presencia de agregados o acúmulos de proteínas anómalas a nivel de las neuronas o la glía [[25]] cuya identificación ha contribuido al conocimiento de los mecanismos patogénicos, además de facilitar la clasificación de este tipo de demencias. El grupo de Cairns establece 4 grupos principales [23] (Tabla 2):
1. En más del 40% de los casos las inclusiones contienen formas insolubles e hiperfosforiladas de la proteína Tau dando lugar a las denominadas Taupatías [10]. 2. Demencia sin cambios histológicos específicos (DLDH) [[27]] cuya denominación viene determinada por la carencia de un fenotipo histológico distintivo. 3. La reevaluación posterior de muchos de los casos incluídos en el grupo anterior ha puesto de manifiesto cómo en su mayoría presentaban inclusiones Ubiquitina-positivas (UBI) siendo denominadas DLFT Ubiquitina-positivas (DLFT-U) [[28],[29]], con una representación superior al 50% del total de las demencias frontales. En los últimos años se han descrito varios subtipos de DLFT-U tras el reconocimiento de la proteína TDP-43 como el principal integrante de las UBI [13], así como algunas formas de DLFT-U TDP-43 negativas [23]. 4. Demencia con acúmulo de filamentos intermedios (NIFID) [[30],[31]]: cuarto grupo muy esporádico caracterizado por la presencia de inclusiones de neurofilamentos alfa-internexina positivas. Basándonos en la clasificación histopatológica de Cairns y colaboradores pasaremos a describir las características histológicas, genéticas y clínicas específicas de los distintos tipos de DLFT. El citoesqueleto celular formado por microtúbulos, microfilamentos y filamentos intermedios constituye un elemento esencial para el mantenimiento de la estructura neuronal (diapositiva 3). El ensamblaje y estabilización de los microtúbulos se ve favorecido por una proteína soluble, la proteína Tau y en concreto por el grado de fosforilación de la misma [[32]]. Las taupatías son un grupo de enfermedades en las que esta proteína aparece hiperfosforilada formando parte de agregados insolubles lo que permite su detección por métodos bioquímicos e histopatológicos. Estos agregados se acumulan a nivel de las neuronas o las células gliales conformando depósitos o inclusiones con una morfología típica según las distintas enfermedades. Hablaremos de Taupatías primarias si el mecanismo inicial es la mutación del gen de la proteína Tau, con lo que ésta será producida de forma anómala (DLFT ligada al cromosoma 17 p.ej). Ahora bien, si tras la producción de una proteína Tau normal ésta se ve alterada por una serie de mecanismos secundarios estaremos ante las Taupatías secundarias (EA p.ej.) en las que de igual forma se producirá una hiperfosforilación y agregación en acúmulos [32] (diapositiva 4). La proteína Tau se encuentra en el cerebro adulto en seis isoformas diferentes en función de la inclusión o exclusión de ciertos exones durante el procesamiento del RNA mensajero. Tres de ellas con una carencia del exón 10 del gen Tau presentan tres repeticiones de una región de unión a microtúbulos (Tau 3R). Las otras tres isoformas que sí incluyen el exón 10 contienen cuatro repeticiones de esta región (Tau 4R) [[33]]. Según el predominio de cada una de estas isoformas se ha establecido la siguiente clasificación de las Taupatías [23] (Tabla 3):
SÍNDROME CORTICOBASALEn 1967 Rebeitz y colaboradores [[34]] describieron la denominada “degeneración corticodentadonígrica con acromasia” en varios pacientes con un síndrome rígido-acinético y apraxia. Las descripciones posteriores de nuevos casos han permitido definir un cuadro clínico, la Degeneración Corticobasal (DCB), caracterizado por rigidez y apraxia asimétricas progresivas con signos de afectación cortical (fenómeno del miembro ajeno, alteración sensitiva cortical, mioclonías o movimientos especulares) y de los núcleos de la base (distonía, temblor o bradicinesia). No obstante la tendencia actual es referirse a esta enfermedad como Síndrome Corticobasal debido a la existencia de casos con manifestaciones parecidas en las que el estudio neuropatológico posterior no ha permitido su confirmación. Es decir, el conjunto de síntomas clínicos descritos no es específico de la DCB por lo que es más adecuado hablar de Síndrome [[35]]. SÍNDROME PARÁLISIS SUPRANUCLEAR PROGRESIVAEn 1964 Steel y colaboradores describen por primera vez un cuadro clínico caracterizado por rigidez y acinesias simétricas con empeoramiento progresivo, parálisis supranuclear de la mirada y trastorno de la marcha severo al que denominaron Parálisis Supranuclear Progresiva (PSP) [[36]]. Al igual que ocurre con la DCB se tiende a utilizar el término Síndrome de Parális Supranuclear Progresiva debido a que se ha podido comprobar cómo un pequeño porcentaje de pacientes con sintomatología propia de la PSP tienen enfermedades con sustrato histopatológico diferente [[37]]. DLFT-17 CON PARKINSONISMOEn 1994 se describió por primera vez una enfermedad familiar ligada al cromosoma 17q21 caracterizada por desinhibición, demencia, parkinsonismo y amiotrofia [[38]] lo que posteriormente se denominaría como Demencia frontotemporal ligada al cromosoma 17 con parkinsonismo (DLFT-17) (diapositiva 5). El gen MAPT encargado de la codificación de la proteína asociada a los microtúbulos es localizado en este cromosoma en el año 1998 estableciéndose su relación con la DLFT-17 [10]. Cualquier mutación de este gen determina la formación de agregados insolubles de proteína Tau hiperfosforilada en las neuronas y en la glía en forma de patología Tau 3R, 4R o 3R/4R. Hasta la fecha se han descrito 41 mutaciones en más de 100 familias [[39]] de modo que las DLFT-17 suponen del 5 al 10% de las formas hereditarias de la DLFT. Las mutaciones del gen MAPT pueden dividirse en dos grupos dependiendo del mecanismo primario implicado: en el primero se produce una alteración del balance normal de ensamblaje del exón 10 con un incremento del mismo, lo que determina un aumento en la ratio 4R/3R tau [24]; el segundo mecanismo determina una alteración en la función de la proteína tau con la disminución en su capacidad para unirse a los microtúbulos favoreciendo así su agregación [39]. El depósito de proteína Tau constituye el principal marcador histopatológico de las DLFT por mutaciones del gen MAPT con una morfología diferente en función del depósito de proteína Tau 3R/4R, 4R o 3R. Clínicamente los pacientes presentan un trastorno conductual típico de la DFT al que suele asociarse un cuadro de parkinsonismo. Sin embargo, el espectro es muy amplio pudiendo presentar síntomas similares a la Esclerosis Lateral Amiotrófica, Degeneración Corticobasal o Parálisis Supranuclear Progresiva con una marcada variabilidad intra e interfamiliar [39].
UBIQUINOPATÍAS: HISTOPATOLOGÍA Y GENÉTICA DE LAS DLFT-U El principal sistema de degradación de proteínas de las células (diapositiva 6) es el denominado Proteosoma. Ahora bien, para que una proteína anómala sea reconocida y degradada ha de ser marcada previamente con Ubiquitina. De existir fallos en este sistema se producirán diferentes enfermedades neurodegenerativas con un denominador común: el excesivo acúmulo de proteínas ubiquitinizadas servirá como marcador al presentar una reacción inmunohistoquímica altamente positiva a la Ubiquitina. Es precisamente esta característica la que ha permitido diferenciar las DLFT en dos grandes subgrupos: el primero con inclusiones Tau-positivas o Taupatías (ver apartado anterior) y el segundo con inclusiones Ubiquitina-positivas (UBI), Tau y a-sinucleína- negativas [28] (DLFT-U) como se describirá a continuación.
BIOLOGÍA Y PATOLOGÍA DE LA TDP-43La identidad de la proteína acumulada en las inclusiones Ubiquitina-positivas (UBI) de las DLFT-U ha permanecido desconocida hasta hace poco tiempo, a pesar de los estudios exhaustivos realizados utilizando técnicas similares a las empleadas en otras enfermedades neurodegenerativas como las taupatías o las sinucleopatías. Es en el año 2006 cuando Neummann y colaboradores [13] indentificaron la TDP-43 como la principal proteína acumulada en estas inclusiones. La TDP-43 (proteína fijadora del ADN TAR 43) es una proteína nuclear localizada en múltiples tejidos del organismo incluyendo el corazón, pulmón, bazo, riñón, músculo o cerebro [[40]] codificada por el gen TARDBP localizado en el cromosoma 1. Responsable de múltiples funciones está implicada en mecanismos reguladores tanto de la transcripción del mRNA como del ensamblaje de diversas proteínas. El secuestro de la TDP-43 en inclusiones ubiquitinadas citoplasmáticas o intranucleares produce una pérdida de la función de esta proteína lo que en última instancia se traduce en una alteración de la transcripción del mRNA y un ensamblaje aberrante del pre-mRNA. Recientemente se ha puesto de manifiesto cómo la alteración patológica de la TDP-43 juega un papel muy relevante en la disfunción neuronal de un grupo heterogéneo de enfermedades como son la DLFT-U esporádica y familiar con y sin enfermedad de motoneurona (EMN) asociada, así como en formas familiares y esporádicas de Esclerosis Lateral Amiotrófica (ELA). La bioquímica en estos desórdenes muestra como la TDP-43 es anormalmente fosforilada, ubiquitinada y dividida para generar fragmentos C-terminales y cómo sólo se detectan las inclusiones con inmunoreactividad positiva para la Ubiquitina en algunas zonas del cerebro como el hipocampo, el neocórtex y la médula espinal. La principal característica neuropatológica de estas enfermedades viene determinada la presencia de inclusiones neuronales citoplasmáticas (NCIs), inclusiones neuronales intranucleares (NIIs), neuritas distróficas (DNs) e inclusiones citoplasmáticas gliales (GCIs) cuyo rasgo distintivo es el de presentar inmunoreactividad positiva para la Ubiquitina y la proteína TDP43, siendo negativas para Tau, α-sinucleína, β-amiloide, filamentos neuronales intermedios y poliglutaminas. La variabilidad de los tipos morfológicos de las inclusiones, su distribución, densidad y perfil inmunohistoquímico ha llevado a proponer una clasificación de las DLFT-U en cuatro subtipos patológicos [[41],[42],[43]] que a su vez responden a dos modelos histológicos de DLFT-U: el elaborado por Sampathu y colaboradores [43] basándose únicamente en la distribución de la patología cortical y el elaborado por Mackenzie y colaboradores [[44]] considerando las inclusiones tanto a nivel cortical como a nivel de la fascia dentada. La proteína TDP-43 constituye por tanto la principal proteína anómala localizada en las UBI características de casi todos los subtipos de DLFT-U [13] siendo negativa su detección en algunos tipos de DLFT-U (ver tabla 2), en otros tipos de DLFT (DFT con cuerpos de Pick, DCB, PSP, DLFT-17 con mutaciones del gen MAPT, enfermedad con inclusiones neuronales de filamentos intermedios, demencia con cuerpos de inclusión basófilos, demencia con granos argirófilos) u otras demencias degenerativas (EA, Enfermedad de Parkinson, Demencia con cuerpos de Lewy, Atrofia Multisistémica) [41]. Este hecho ha permitido establecer una nueva clasificación de las DLFT-U dando lugar a las denominadas TDP-43 proteinopatías o TARDopatías [[45]] en las que parece existir una correlación clínico-patológica (diapositiva 7) (Tabla 4).
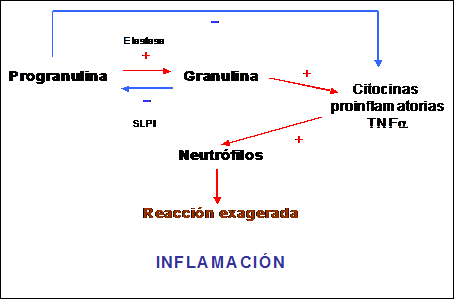
PROGRANULINA Y TDP-43Muchos son los artículos publicados, tras la descripción original de las mutaciones del gen MAPT, sobre familias con DLFT y herencia autosómica dominante, en las que a pesar de estar ligadas genéticamente a la misma región del cromosoma 17q21 que contiene el gen MAPT no se pudo encontrar ninguna de las mutaciones descritas para el mismo [[46]]. En estas familias se pudo constatar la presencia de una proteína Tau normal además de inclusiones Ubiquitina/TDP-43 positivas. La ausencia de mutaciones del gen MAPT y la presencia de una histopatología DLFT-U apuntaba a la existencia de un segundo gen en la misma región del cromosoma 17. En el año 2006 Baker y colaboradores demostraron cómo el gen de la Progranulina (PGRN) situado a 1.7Mb del gen MAPT era el responsable de este subtipo de DLFT (diapositiva 8) [11]. La PGRN [[47]] es una proteína con un peso molecular de 68.5 kDa codificada por un único gen localizado en el cromosoma 17q21, expresada en múltiples tejidos e implicada en diversas funciones. A nivel periférico se encuentra presente en las células epiteliales (piel, tracto gastrointestinal y epidídimo) y en las células hematopoyéticas desarrollando un importante papel en los procesos inflamatorios, de curación de las heridas o de la tumorogénesis. En condiciones normales existe un equilibrio entre la PGRN y su principal metabolito, la Granulina. Este péptido es liberado por la acción proteolítica de la Elastasa, enzima cuya acción se encuentra a su vez inhibida por la Proteasa Inhibitoria de la secreción de Leucocitos (SLPI) [[48]]. Si este equilibrio se altera en favor de la producción de Granulina se desencadena una reacción inflamatoria debido a la liberación de sustancias proinflamatorias (citocinas y TNFα) (Figura 1). Si este proceso adquiere una intensidad adecuada será útil ante cualquier tipo de herida donde además la PGRN, debido a sus propiedades tróficas, activa la producción de fibroblastos, células endoteliales, macrófagos y neutrófilos. Mediante un efecto paracrino favorecerá así mismo al proceso de epitelización, granulación y vascularización de las heridas. Sin embargo, este mismo efecto trófico presenta su expresión más negativa en la tumorogénesis donde la presencia de PGRN favorece su desarrollo y crecimiento [[49]].
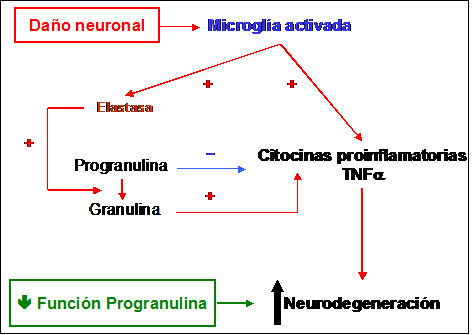
A nivel del Sistema Nervioso Central (SNC) la PGRN se encuentra en las neuronas piramidales, neuronas del hipocampo y células cerebelosas de Purkinje, si bien se ha podido comprobar una pérdida progresiva en la expresión de esta proteína en el cerebro humano desde su etapa de embrión (células neuroepiteliales) y feto (tronco del encéfalo, bulbo olfatorio, retina y médula espinal) a la de adulto, lo que sugiere un papel como factor estimulante del desarrollo y diferenciación del SNC. Es de especial relevancia la presencia de PGRN a nivel de la microglía debido a que estas células se activan ante cualquier situación que induzca un posible daño neuronal: traumatismos, infecciones, etc. La consecuencia es la liberación por una parte de sustancias proinflamatorias (citocinas y TNFα) y por otra de Elastasa, enzima que favorecerá la producción de Granulina en detrimento de la PGRN incrementando la intensidad del proceso inflamatorio y favoreciendo así la neurodegeneración. Si por cualquier motivo la función o la producción de PGRN se ve disminuída también lo hará su efecto inhibitorio sobre la inflamación y se verá favorecido por tanto el proceso neurodegenerativo [49] (Figura 2).
En el último año han sido descritas más de 44 mutaciones del gen de la PGRN en más de 82 familias [[50],[51]]. En todas ellas se producen alelos nulos dando lugar a una terminación prematura de la secuencia de codificación con la producción de un mRNA mutado sin capacidad para producir una proteína funcional [[52]]. Como consecuencia no se produce una proteína mutada ni un acúmulo de la misma a nivel de las inclusiones neuronales sino una disminución de su función o Haploinsuficiencia lo que a su vez condiciona un aclaramiento anormal, ubiquitinización y acúmulo de la proteína TDP43 en las inclusiones neuronales. Todas las familias con mutaciones del gen de la PGRN presentan un patrón neuropatológico típico caracterizado por una marcada atrofia a nivel frontal, núcleo caudado, sustancia negra y región medial del tálamo. Todas muestran además un subtipo histológico característico con afectación predominante de las capas corticales más superficiales del neocórtex frontotemporal y del estriado con la presencia de abundantes inclusiones neuronales intracitoplasmáticas (NCIs) Ubiquitina/TDP43 positivas. Sin embargo, son las inclusiones neuronales intranucleares (NIIs) su principal característica debido a su forma lenticular o en ojos de gato (“cat eyes”) [[53], [54]]. De acuerdo a la clasificación histopatológica de las TDP-43 proteinopatías, la DLFT-U por mutación del gen de la PGRN se correspondería con el subtipo 3 de Sampathu [43] o el subtipo 1 de Mackenzie [44].
FENOTIPO CLÍNICO EN LAS PROGRANULOPATÍASLa DLFT-U por mutación de la PGRN representa algo más del 50% del total de DLFT y hasta un 60% de las forma hereditarias. Con un transmisión autosómica dominante y una elevada penetrancia sitúa en torno a los 60 años la edad de inicio de los síntomas. El fenotipo clínico es normalmente una combinación de trastorno conductual y de la personalidad con afectación del lenguaje en forma de Afasia Primaria Progresiva No Fluente. Un cuadro de parkinsonismo asociado es muy frecuente ya desde las etapas iniciales siendo excepcional la asociación de una enfermedad de motoneurona [52]. Cabe destacar una marcada variabilidad no sólo entre familias sino entre los componentes de una misma familia, circunstancia ésta que dificulta enormemente el predecir cuál de las formas familiares de DLFT tendrá como causa una mutación del gen de la PGRN [[55]]. No obstante, en aquellos casos de DLFT-U sin mutaciones del gen de la PGRN la prevalencia de trastorno del lenguaje o parkinsonismo es mucho menor [[56]] lo que sí permitiría aventurar un diagnóstico diferencial entre estos dos subtipos de DLFT-U. GEN VCP - CROMOSOMA 9 – TDP-43 La Miopatía con cuerpos de inclusión asociada a Enfermedad de Paget ósea y Demencia Frontotemporal (IBMPFD) es una rara enfermedad autosómica dominante causada por una mutación del gen VCP (valosin-containing protein) localizado en el cromosoma 9p21-12 [[57]]. El VCP forma parte de la superfamilia de genes AAA-ATPasa (ATPasa asociada con diversas actividades celulares) [[58]] cuyas funciones son múltiples incluyendo su participación en la degradación de las proteínas mediante su interacción con el sistema ubiquitina-proteosoma, en la reparación del ADN, en la respuesta al estrés o en la muerte celular programada. El patrón neuropatológico de este tipo de demencia muestra una importante afectación de las regiones corticales del neocórtex siendo menor a nivel subcortical, junto a la presencia en estas localizaciones de numerosos NIIs y relativamente escasos NCIs y DNs, Ubiquitina/TDP-43 positivas y VCP negativas [[59]]. De acuerdo a la clasificación histopatológica de las TDP-43 proteinopatías se correspondería con el subtipo 4 de Sampathu [43] y Mackenzie [44]. En la biopsia muscular de forma típica aparecen los cuerpos de inclusión miopáticos. Clínicamente la IBMPFD se caracteriza por un cuadro de debilidad en individuos adultos semejante al que aparece en la distrofia de las cinturas, una Enfermedad de Paget ósea que se presenta con una penetrancia variable y una demencia frontal que afecta al 30% de los pacientes con un inicio precoz. ESCLEROSIS LATERAL AMIOTRÓFICA Y TDP-43 La Esclerosis Lateral Amiotrófica (ELA) es una enfermedad neurodegenerativa en la que como consecuencia de la afectación de las neuronas motoras se produce una debilidad progresiva llegando a la parálisis y muerte del paciente en unos 3 a 5 años [[60]]. La mayor parte de los casos de ELA son esporádicos, si bien un 10% presentan una historia familiar positiva de los cuales en torno a un 20% son debidos a una mutación en el gen de la Superóxido Dismutasa (SOD1) [60,[61]]. Clínicamente un 20% de casos de ELA asocian una demencia de tipo frontal y en torno al 50% presentarán deterioro cognitivo en mayor o menor grado a lo largo de su evolución [[62]]. Los hallazgos neuropatológicos incluyen una marcada pérdida de neuronas motoras superiores e inferiores, degeneración de los tractos corticoespinales, cuerpos de Bunina [[63]] y la presencia de inclusiones Ubiquitina-positivas en el citoplasma de las neuronas motoras dañadas. Tanto en las formas de ELA esporádicas como familiares SOD1 negativas estos NCIs presentan típicamente forma de madeja (skein-like inclusions), siendo TDP-43 positivas [13]. En definitiva, tanto las formas de ELA esporádica como familiares formarían parte de las TDP-43 proteinopatías o Tardopatías, exceptuando aquellas formas familiares por mutación del gen SOD1, en cuyas inclusiones se encuentra de forma característica SOD1 [[64]]. ENFERMEDAD DE MOTONEURONA Y TDP-43Las enfermedades que afectan a la motoneurona (EMN) son clínica y patogénicamente muy heterogéneas. Suelen clasificarse de acuerdo a la afectación de la primera motoneurona, de la segunda o de ambas siendo la ELA la forma más frecuente. Algunas formas de EMN afectan inicialmente a las neuronas de la médula espinal y del tronco del encéfalo, mientras que en otras se produce una afectación en todos los niveles del neuroeje. A pesar de los avances genéticos la posible asociación entre las distintas formas de EMN ha permanecido sin aclarar hasta que Neumann y colaboradores [13] demostraron la presencia de la proteína TDP-43 no sólo en las UBIs de la DLFT-U sino también en las inclusiones neuronales de la ELA. Posteriormente estas misma inclusiones se han descrito en la DLFT asociada a EMN (DLFT/EMN), en algunos casos de Esclerosis Lateral Primaria y en las neuronas motoras de hasta un 10% de las DLFT en las que no había síntomas clínicos o afectación neuropatológica sugestiva de EMN [[65]]. Estos hallazgos parecen confirmar que la DLFT-U asociada a EMN y la ELA formarían parte del mismo espectro clínico-patológico de una enfermedad degenerativa ligada a la patología de la proteína TDP-43 [13]. De igual modo la presencia de inclusiones TDP43-positivas permitiría establecer el diagnóstico diferencial de la ELA y DLFT/EMN de aquellas otras enfermedades de motoneurona o de DLFT con formas subclínicas de EMN. CROMOSOMA 9 - DLFT-EMN – TDP-43 Han sido descritas varias familias con miembros afectos de DLFT, EMN o DLFT/EMN con herencia ligada al Cromosoma 9p21-13 [[66],[67],[68]]. Todas ellas presentaban un patrón neuropatológico correspondiente a la DLFT-U con abundantes NCIs y escasos DNs/NIIs de acuerdo al subtipo histológico tipo 2 de Sampathu [43] o 3 de Mackenzie [44]. En una única familia con herencia ligada al cromosoma 9p se ha implicado a una mutación del gen de la proteína 74 para el transporte intraflagelar (IFT74) como responsable [67]. La IFT74 es una proteína localizada en el compartimento vesicular intracelular y forma parte del sistema de transporte intraflagelar implicado en el transporte vesicular de material sintetizado desde el soma neuronal hacia las dendritas y el axón. FORMAS NEGATIVAS DE TDP-43- DLFT ligada al Cromosoma 3 -La DLFT con mutación del gen Charged multivesicular body protein 2B (CHMP2B) es la causa de la DFT ligada al cromosoma 3 detectada en una única familia danesa [[69], [70]]. Este gen codifica un componente del complejo secretor endosómico tipo III (ESCRTIII) necesario para la formación de cuerpos multivesiculares implicados en la degradación de las proteínas. El patrón neuropatológico característico está constituido por la presencia de abundantes NCIs Ubiquitina positivas limitados al hipocampo y en las que a pesar del empleo de diferentes técnicas inmunohistoquímicas no se ha podido demostrar reactividad frente a la proteína TDP-43, por lo que esta enfermedad formaría parte de las DLFT-U pero no de las TDP-43 proteinopatías [41]. Clínicamente la DLFT-U ligada al Cromosoma 3 se caracteriza por un inicio de los síntomas en la sexta década de la vida con cambios en la personalidad y el comportamiento, así como por una afasia progresiva. - Enfermedad con Cuerpos de Inclusión Basófilos (BIDD))Enfermedad caracterizada por una sintomatología atípica al inicio con debilidad, disartria y pérdida de memoria. La demencia sobreviene en torno al año de evolución asociando signos de afectación de las neuronas motoras superior e inferior, parkinsonismo, síntomas parietales y movimientos anormales. Existe una severa atrofia de la corteza, ganglios basales, sustancia negra y tractos piramidales, con mínima afectación de la motoneurona inferior. Las inclusiones neuronales son típicamente negativas para tau, α-sinucleína, α-internexina y neurofilamentos, pero positivos para Ubiquitina y la Proteína P62 [[71]]. DEMENCIA SIN CAMBIOS HISTOLÓGICOS ESPECÍFICOS (DLDH) La clasificación histopatológica de las DLFT se realiza atendiendo a la composición histobioquímica de las inclusiones neuronales cerebrales [23]. En la denominada Demencia sin cambios histológicos específicos (DLDH) no se ha podido determinar mediante diferentes técnicas inmunohistoquímicas cambios neuropatológicos típicos, siendo su principal característica la ausencia de depósitos anómalos de proteína Tau, inmunoreactividad positiva frente a la Ubiquitina o la presencia de cualquier otro tipo de inclusión neuronal [27]. Hasta fechas recientes se consideraba que una proporción importante de DLFT carecían de un fenotipo histológico distintivo, pero muchos de estos casos pasaron a formar parte del grupo de DLFT-U gracias al descubrimiento de genes como el de la Progranulina [11] o al desarrollo de nuevas técnicas que en última instancia han llevado al descubrimiento de la proteína TDP-43 [13] como principal elemento integrante de las UBIs. Presumiblemente el porcentaje de casos de DLDH continuará disminuyendo en la medida en que aparezcan nuevas proteínas o genes responsables de las inclusiones neuronales en las DLFT. ENFERMEDAD POR INCLUSIÓN NEURONAL DE FILAMENTOS INTERMEDIOS (NIFID). Los filamentos intermedios (IF) están compuestos por tres subunidades protéicas fosforiladas denominadas light (NL-F), medium (NL-M) y heavy (NL-H). Existe además una cuarta proteína integrante de estos neurofilamentos, la α-internexina, codificada por un gen localizado en el cromosoma 10. Esta proteína es expresada por la práctica totalidad de las neuronas en su etapa de diferenciación de forma previa al triplete de proteínas anterior lo que favorece su ensamblaje, pero con un descenso muy significativo de su presencia en el cerebro adulto. Su sobreexpresión se traduce en un acúmulo anormal de los neurofilamentos dando lugar a las inclusiones α-internexina positivas características de la NIFID. La enfermedad por inclusión de filamentos intermedios es un enfermedad neurodegenerativa muy infrecuente caracterizada por una importante variabilidad clínica en la que se incluye una demencia de perfil frontotemporal de inicio precoz, junto a la presencia de signos piramidales y extrapiramidales (diapositiva 14). Macroscópicamente se pone de manifiesto una marcada atrofia de los lóbulos frontales y en menor grado de los temporales y parietales. A nivel microscópico la característica fundamental es la presencia de inclusiones neuronales de filamentos intermedios Tau y α-sinucleína negativos [30,31,72]. Si bien la presencia de inclusiones α-internexina positivas es el marcador histopatológico de este tipo de demencia, se han podido encontrar pequeños acúmulos de esta proteína en las inclusiones neuronales de otras enfermedades neurodegenerativas como la EA, la demencia con cuerpos de Lewy o la EMN [[73]].
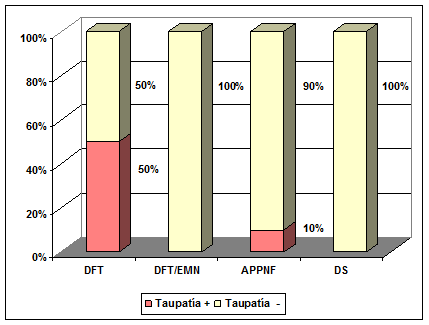
¿Puede hablarse de una correlación clínico-patológica en las Demencias Lobares Frontotemporales?La búsqueda de tratamientos etiológicos en la DLFT supone la necesidad de desarrollar biomarcadores que permitan un diagnóstico lo más exacto posible de la enfermedad subyacente. Sin embargo, la heterogeneidad clínica y neuropatológica, así como la inestabilidad evolutiva de los distintos síndromes han supuesto una enorme dificultad para el logro de este objetivo. Tradicionalmente se ha considerado a las DLFT un grupo heterogéneo de enfermedades neurodegenerativas en las que resultaba extraordinariamente difícil establecer una relación entre la presentación clínica y el proceso patológico subyacente. Tampoco era posible conociendo el diagnóstico patológico deducir la naturaleza de las manifestaciones clínicas. En el momento actual y gracias a los avances en genética molecular, bioquímica y neuropatología de las DLFT se puede aproximar no sólo una correlación clínico-patológica, sino también clínica, histológica, genética y biomolecular. Las formas Tau-positivas se suelen asociar a la presencia de síntomas extrapiramidales hasta en dos tercios de los casos, a síndromes como la Degeneración Corticobasal o a un trastorno del lenguaje en forma de Afasia Primaria Progresiva No Fluente con una marcada afectación de las funciones visuoconstructivas. La patología Tau-negativa presenta con mayor frecuencia afectación del comportamiento y del lenguaje con una mayor dificultad en la nominación [[74]]. Snowden y colaboradores [[75]] examinaron los cerebros de 79 pacientes seguidos longitudinalmente a lo largo de su vida y en los que el análisis postmortem evidenció una DLFT según criterios histopatológicos. Estos autores han podido comprobar cómo el fenotipo clínico no sólo tiene una relación predecible con la presencia o ausencia de patología Tau, sino que en el grupo de DLFT-U también es posible establecer una relación entre el fenotipo clínico y el subtipo histológico de TDP-43. Los pacientes con Taupatía representaron el 38% de la muestra y en el 62% restante se constató ausencia de patología Tau con una distribución fenotípica muy característica como puede apreciarse en el gráfico 3 y la tabla 5.
Finalmente en la Tabla 6 quedan reflejadas las correlaciones histopatológicas, fenotipos clínicos, cromosomas y genes implicados en las distintas formas de DLFT Ubiquitina positivas (DLFT-U).
Evolución de las Demencias Lobares FrontotemporalesLos síndromes clínicos agrupados en el concepto de DLFT se caracterizan por su inestabilidad debido a los cambios desarrollados a lo largo de la evolución de la enfermedad. De forma habitual suele ser un solo síndrome el que predomina durante los 1 a 4 años iniciales evolucionando posteriormente a otro [[76]]. Kertesz y colaboradores [[77]] constataron la aparición de un segundo e incluso un tercer síndrome en un estudio prospectivo de 262 pacientes con criterios clínicos de DLFT: la DFT desarrolló hasta en un 50% de los casos una APPNF, un 22.5% presentó un síndrome de DCB/PSP y en torno al 16% una DS. Algo más del 50% de las APPNF desarrollaron una DFT, un tercio un síndrome DCB/PSP y el resto no evolucionó a ningún otro cuadro clínico. La DS desarrolló como segundo síndrome una DFT en tres cuartas partes de los casos o ningún otro cuadro clínico en el grupo restante. Finalmente los casos con el síndrome DCB/PSP presentaron como segundo síndrome una DFT (50%) o una APPNF (50%) (tabla 7).
Este solapamiento entre los distintos síndromes clínicos característicos de la DLFT nos lleva a considerar que realmente no sean entidades independientes entre sí, teniendo en cuenta la posible aparición de diferentes expresiones clínicas en un mismo paciente a medida que la enfermedad progresa. Podría afirmarse que un mismo tipo histopatológico podría generar la aparición de distintos cuadros clínicos por mecanismos etiológicos o neuropatológicos no completamente clarificados en el momento actual. Además, esta variabilidad fenotípica puede ocurrir tanto entre pacientes diferentes como en un mismo paciente a lo largo de la evolución de la enfermedad. Por otra parte, esta heterogeneidad hace imprescindible la búsqueda de biomarcadores que permitan un diagnóstico etiológico lo más preciso posible a fin de poder desarrollar tratamientos específicos.
ResumenEn el término Demencia Lobar Frontotemporal (DLFT) se agrupan un conjunto de enfermedades neurodegenerativas muy heterogéneo en su expresión clínica, componente genético y características histopatológicas, lo que tradicionalmente ha dificultado su estudio y clasificación. Los pacientes presentan de forma habitual un cambio progresivo en su conducta asociado a alteración del lenguaje y pérdida de memoria, constituyendo la segunda causa de demencia en personas menores de 65 años. La característica más relevante a nivel histopatológico es la presencia de agregados o acúmulos de proteínas anómalas a nivel de las neuronas o la glía, cuya identificación ha contribuido por una parte al conocimiento de los mecanismos patogénicos y por otra ha permitido la clasificación de este tipo de demencia. En las dos últimas décadas se ha producido un notable progreso en el conocimiento de este grupo de enfermedades gracias a los avances genéticos relacionados con el descubrimiento del gen MAPT y el gen de la Progranulina, así como de sus mutaciones responsables de un porcentaje elevado de las DLFT hereditarias; igualmente, el desarrollo de nuevas técnicas inmunohistoquímicas ha permitido caracterizar algunas proteínas anómalas como la proteína TDP-43 como principal integrante de las inclusiones neuronales en las DLFT tau negativas. Todo ello ha permitido establecer una nueva clasificación de las DLFT. Con el siguiente trabajo hemos pretendido realizar una revisión exhaustiva de dichos avances, además de considerar las posibles posibles correlaciones clínicas, histológicas, genéticas y biomoleculares de los distintos subtipos de DLFT.
[1] - Ikeda M, Ishikawa T, Tanabe H. Epidemiology of frontotemoral lobar degeneration. Dement Geriatr Cogn Disord 2004; 17: 265-268. [2] - **Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S et al . Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998; 51: 1546-1554. [3] - Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal demencia. Neurology 2002; 59: 1077-1079. [4] - Serra-Mestres J. Síndromes degenerativos focales. En: López-Pousa, Vilalta FranchJ, Llinás Reglá J (Eds.) Manual de Demencias. Barcelona: Prous Science, 1996; 374-383. [5] - Brun A. Frontal lobe degeneration of non-Alzheimer type. I. Neuropathology. Arch Gerontol Geriatr 1987; 6:193-208. [6] - Gustafson L. Frontal lobe degeneration of non-Alzheimer type. I. Clinical picture and differential diagnosis. Arch Gerontol Geriatr 1987; 6: 209-223. [7] - **Neary D, Snowden JS, Northern B and Gouldin P. Dementia of frontal lobe type. J Neurol Neurosurg Psychiatry 1988; 51: 353-361. [8] - Mesulam M. Slowly progressive aphasia without dementia. Ann Neuolo 1982; 11: 592-598. [9] - Snowden JS, Goulding PJ, Neary D. semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol 1989; 2: 167-82. [10] - **Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al. Association of missense and 5´-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998; 393: 702-705. [11] - **Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 916-919. [12] - **Cruts M, Gijselinck I, Van Der Zee J, Engelborghs S, Wils H, Pirici D et al. Null mutations in progranulin cause ubiquitine-positive frontotemporal dementia linked to chromosome 17. Nature 2006; 442: 920-924. [13] - **Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi Mc, Chou TT et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006; 314: 130-133. [14] - Alonso-Navarro H, Jabbour-Wadih T, Ayuso-Peralta L, Jiménez-Jiménez F.J. Neuroquímica y neurofarmacología de la demencia frontotemporal. Rev Neurol 2006; 42:556-561. [15] - Mega M, Cummings JL. Frontal-Subcortical Circuits and Neuropsychiatric Disorders. Journal Of Neuropsychiatry Clin Neurosci 1994; 6: 358-370. [16] - Serra Mestres J. Síndromes degenerativos focales. En: Manual de demencias. López Pousa S, Vilalta Franch J, Llinás Reglá J (Eds). Prous Science Ediciones, 2001. [17] - O´Neill S, Andreotti M, De Simone V. Demencia semántica, una enfermedad de muchas palabras. Rev Neurol 2006; 43: 685-689. [18] - ** Gorno-Tempini ML, Dronkers NF, Rankin DP, Ogar JM, Phengrasamy L, Rosen et al. Cognition and anatomy in three variants primary progressive aphasia. Ann Neurol 2004; 55: 335-346. [19] - Serrano C, Martelli M, Haris P, Tufró G, Ranalli C, Taragano F et al. Afasia progresiva primaria: variabilidad clínica. Análisis de 15 casos. Rev Neurol 2005; 41: 527-530. [20] - **Lund, Manchester groups. Consensus statement. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry 1994; 57: 416-418. [21] - Knopman DS, Boeve BF, Parisi JE, Dickson DW, Smith GE, Ivnik RJ et al. Antemortem diagnosis of frontotemporal lobar degeneration. Ann Neurol 2005; 57: 480-488. [22] - **Mackhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia. Report of the Work Group on frontotemporal dementia and Pick´s disease. Arch Neurol 2001; 58: 1803-1809. [23] - **Cairns NJ, Bigio EH, Mackenzie IRA, Hatanpaa KJ, White CL, Schneider JA et al. Neuropathological diagnostic criteria and nosology of the frontotemporal lobar degenerations: consensus criteria of the Consortium for frotntotemporal lobar degeneration. Acta Neuropathol 2007; 114: 5-22. [24] - Neary D, Snowden JS, Mann DD. Frontotemporal dementia. Lancet Neurology 2005; 4: 771-779.Trojanowski JQ, Dickson D. Update on the neuropathological diagnosis of frontotemporal dementia. J Neuropathol Exp Neurol 2001; 60: 1123-1126. [25] - Trojanowski JQ, Dickson D. Update on the neuropathological diagnosis of frontotemporal dementia. J Neuropathol Exp Neurol 2001; 60: 1123-1126. [26] - **Toribio-Díaz, ME; Morera-Guitart J. Clasificación clínica y biomolecular de las demencias frontotemporales. Revisión de la bibliografía.Rev Neurol 2008; 47: 588-598. [27] - **Mann DM, McDonagh AM, Snowden J, Neary D, Pickering-Brown SM. Molecular classification of the dementias. Lancet 2000. 355: 626. [28] - Josephs KA, Holton JL, Rossor MN, Godbolt AK, Ozawa T, Strand K et al. Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 2004. 30: 369-373. [29] - Mackenzie IR, Shi J, Shaw CL, Duplessis D, Neary D, Snowden JS, Mann D. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol 2006. 112: 551-559. [30] - Cairns NJ, Zhukareva V, Uryu K, Zhang B, Bigio E, Mackenzie IR et al. Alpha-internexin is present in the pathological inclusions of neuronal intermediate filament inclusión disease. Am J Pathol 2004; 164: 2153-2161. [31] - Armstrong RA, Kerty E, Skullerud K, Cairns NJ. Neuropathological changes in ten cases of neuronal intermediate filament inclusion disease (NIFID): a study usin alpha-internexin immunohistochemistry and principal components analysis (PCA). J Neural Transm 2006. 113: 1207-1215. [32] - Delacourte A, Buee L. Normal and pathological tau proteins as factors for microtubule assembly. Int Rev Cytol 1997; 171: 167-224. [33] - Delacourte A, Sergeant N, Wattez A, Robitaile Y. The Biochemistry of the Cytosketon in Pick Complex. En: Kertesz A, Muñoz DG (editors). Pick´s Disease and Pick Complex. New York: Wiley-Liss 1998: 243-258. [34] - Rebeitz J, Kolodny E, Richardson E. Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 1967; 92: 23-26. [35] - Boeve B. Corticobasal degeneration: the syndrome and the disease. In: Litvan I, ed. Atypical Parkinsonism Disorders. Totawa: Humana Press; 2005: 309-334. [36] -Steel J, Richardson J, Olszewski J. progressive supranuclear palsy: a heterogeneous degeneration involving the brainstem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 2: 473-486. [37] - Josephs K, Dickson D. Diagnostic accuracy of progressive supranuclear palsy in the Society for Progressive Supranuclear Palsy brain bank. Mov Disord 2003; 18: 1018-1026. [38] - ** Wilhelmsen KC, Lynch T, Pavlou E, Higgins M and Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex 17q21-22. Am J Hum Genet 1994; 55: 1159-1165. [39] - **Rademakers R, Cruts M, Van Broeckhoven C. The role ofl tau (MAPT) in frontotemporal dementia and related taupathies. Hum Mutat 2004; 24: 277-295. [40] - **Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon skipping. EMBO J 2001. 20: 1774-1778. [41] - Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 2007; 171: 227-240. [42] - Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzxchmar HA, Van Deerlin et al. TDP-43 positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 2007; 66: 177-83. [43] - **Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin inmunohistochemistry and novel monoclonal antibodies. Am J Pathol 2006; 169: 1343-1352. [44] - **Mackenzie IR, Baboire A, Pickering-Brown S, Plessis DD, Jaros E, Perry RH et al. Heterogeneity of ubiquitin in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 2006; 112: 539-549. [45] - **Kwong L, Neumann M, Sampathu D, Lee V, Trojanowski J. TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron diseas. Acta Neuropathol 2007. 114: 63-70. [46] - **Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van den Broeck M et al. Tau negativa frontal lobe dementia at 17q21: significant finemapping of the candidate regio to a 4.8 cM interval. Mol Psychiatry 2002; 7: 1064-1074. [47] - He Z, Batemann A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med 2003; 81: 600-612. [48] - Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cel 2002; 11: 867-878. [49] - **Ahmed Z, Mackenzie IRA, Hutton M, Dickson D. Progranulin in frontotemporal lobar degeneration and neuroinflamation. Journal of Neuroinflammation 2007; 4:7 doi: 10.1186/1742-2094-4-7 [50] - Pickering-Brown S. Progranulin and frontotemporal lobar degeneration. Acta Neuropathol 2007; 114: 39-47. [51] - Alzheimers Disease & Frontotemporal Dementia Mutation Database: http://.molgen.ua.ac.be/FTDMutations/: [consulta: 1 Mayo 2008] [52] - Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet 2006; 15: 2988-3001. [53] - Mackenzie IRA, Baker M, Pickering-Brown S, Hsiung GYR, Lindholm C, Dwosh E et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranuline gene. Brain 2006; 129: 3081-3090. [54] - **Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve Bf, Knopman DS et al. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol 2007; 66: 142-151. [55] - Snowden JS, Pickering-Brown SM, Mackenzie IM, Richardson AMT, Varma A, Neary D et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain 2006; 129: 3115-3123. [56] - Mackenzie IAR, Rademakers R. The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 2007; 8: 237-248. [57] - Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D et al. Inclusion body miopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing portein. Nat Genet 2004; 36: 377-381. [58] - Wang Q, Song C, Li CC. Molelcular perspectives on p97-VCP: progress understanding its structure and diverse biological functions. J Struct Biol 2004; 146: 44-57. [59] - Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP mutations. J Neuropathol Exp Neurol 2007; 66: 152-157. [60] - Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 2006; 7: 710-723. [61] - Gros-Louis F,Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta 2006; 1762: 956-972. [62] - Talbot K, Ansorge O. Recent advances in the genetics of ayotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Hum Mol Genet 2006; 15: R182-R-187. [63] - Tomonga M. Selective appearance of Bunina bodies in amyotrophic alteral sclerosis. A study of the distribution in midbrain and sacral cord. J Neurol 1980; 223: 259-267. [64] - Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmatic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropatholo Exp Neurol 1996; 55: 481-490. [65] - Dickson DW, Josephs KA, Amador-Ortiz C. TDP-43 in differential diagnosis of motor neuron disorders. Acta Neuropathol 2007; 114: 71-79. [66] - Morita M, Al Chalabi A, Andersen PM, Soler B, Sapp P, Englund E et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology 2006; 66: 839-844. [67] - Momeni P, Schymick J, Jain S, Cookson MR, Cairns NJ, Gregio E et al. Analysis of IFT74 as candidate for chromosome 9p-linked ALS-FTD. BMC Neurol 2006. doi: 10.1186/1471-2377-6-44. [68] - Valdmanis PN, Dupre N, Bouchard JP, Camu W, Meininger V, Strong M et al. Three families with amyotropic lateral sclerosis and frontotemporal dementia with evidence linkage to chromosome 9p. Arch Neurol 2007; 64: 240-245. [69] - Brown J, Ashworth A, Gydesen S, Sorensen A, Rossor M, Hardy J et al. Familial non-specificic maps to chromosome 3. Hum Mol Genet 1995; 4: 1625-1628. [70] - Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005; 37: 806-808. [71] - Yokota O, Tsuchiya K, Terada S, Ishizu H, Uchikado H, Ikeda M et al. Basophilic inclusion body disease and neuronal intermediate filament inclusion disease: a comparative clinicopathological study. Acta Neuropathol 2008; 115: 561-575. [72] - Molina-Porcel L, Llado A, Rey MJ, Molinuevo JL, Martinez-Lage M, Esteve FX et al. Clinical and Pathological Heterogeneity of Neuronal Intermediate Filament Inclusion Disease. Arch Neurol 2008; 65:272-275. [73] - Cairns NJ, Uryu K, Bigio E, Mackenzie IRA, Gearing M, Duyckaerts C et al. α-internexina aggregates are abundant in neuronal intermediate filament inclusions disease (NIFID) but rare in other neurodegenerative diseases. Acta Neuropathol 2004; 108: 213-223. [74] - Bian H, Grossman M. Frontotemporal lobar degeneration: recent progress in antemortem diagnosis. Acta Neuropathol 114: 23-29. [75] - **Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological realtionships. Acta Neuropathol 2007; 114: 31-38. [76] - Boeve B. Relationship betwee frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy and amyotrophic lateral sclerosis. Alzheimer Dis Assoc Disord 2007; 21: S31-S38. [77] - **Kertesz A, Blair M, McMonagle P, Muñóz D. The diagnostic and evolution of frontotemporal dementia. Alzheimer Dis Assoc Disord 2007; 21: 155-163.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||