Introducción histórica
La Esclerosi Múltiple (EM) definida tal i com avui la definim, val a dir una malaltía d'oritge desconegut caracteritzada per una afectació clínica neurológica diseminada en el temps i l'espai deguda a lesions desmielinitzants aïllades unes d'altres del Sistema Nerviós Central (SNC), preferentment localitzades a la substància blanca (SB) i que es troben en diferents moments evolutius la deguem a Jean-Martin Charcot (1825-1893) (Charcot 1875).
Charcot es va referir a una entitat que afectava al SNC amb exacerbacions i remissions i va reconeixer l'aparició d'una evolució crònica progressiva, aquesta entitat clínica la va relacionar amb una afectació anatòmica del SNC consistent en "la sclerose en plaques disseminées", esclerosi en plaques diseminades. Aquestes plaques serien la marca de la malaltía i estaríen formades per un colecció mixta de cèl.lules: macròfags i astrocits, destrucció de la mielina i preservació de l'axó, a més hi hauría una infiltració perivascular per limfocits, marcròfags i cèl.lules plasmàtiques. Però ja va fer distincions entre una placa activa, amb la presència de macròfags i infiltració perivascular, així com l'activitat en el vessant perifèric d'una placa antiga demostrat per la hipercelularitat del mateix, a aquesta lesió se li anomenà desmielinitzant i des de llavors hom reconeix la Esclerosi en Plaques (EP) o en termionologia moderna Esclerosi Múltiple com una malaltía Desmielinitzant del SNC.
Però Charcot va fer d'altres observacions importants com ara que les fibres desmielinitzades conservaven la capacitat per a conduir l'estimul nerviós o la presència d'alteracions axonals ja en la placa jove d'EM.
Així Charcot va donar les claus per a coneixer aquesta malaltía: Durant la següent exposició intentaré revisar les aportacions que al llarg dels 121 anys que ja dura aquesta investigació, ens han deixat en aquestos tres aspectes: Desmielinitnizació, conducció nerviosa central i dany axonal.
El següent pas en el coneixement de la EM l'hem de situar entre el 1933 i el 1935 amb les publicacions de Rivers i Schwentker (Rivers i cols 1935) d'un model de desmielinització induït experimentalment en monos (EAE), des de llavors aquesta ha estat una tècnica que a condicionat el progrés en el coneixement de la fisiopatogenia i el tractament de la EM.
Però una fita més important en quant a seure les bases patogèniques i d'ajut al diagnòstic d'aquesta malaltía, va ser la demostració de la secreción intratecal d'immunoglobulines duta a terme per Kabat (Kabat i cols 1942) i que índica una activació del sistema immune humoral bé degut a una resposta a un hipotètic virus responsable de la malaltía (Schadlich 1987), bé com a reflexe d'un procés autoimmune.
Des de llavors i fins a la decada dels vuitanta no trobem més referències de caracter fisiopatogènic en la EM, i ja situats als principis dels vuitanta hom admet con a més probable mecanisme fisiopatogènic de la EM, bassat en una alteració del Sistema Immune (SI) mediat per cèlules T i així les definicions a l'abast diuen que "malgrat que la etiopatogènia de la EM no és coneguda existeixen evidències que indiquen que està mediada per cèlules T autorreactives, tot i contribuïnt factors immunogenètics i ambientals 04-Martin R 2/suppl 52), a aquesta conclusió s'arribà fonamentalment per els estudis de la EAE, i més concretament en el model de rata Lewis sensibilitzat per la proteïna bàsica de mielina (PBM) però no hi ha que oblidar que l'anatòmia patològica de les lesions així induïdes no produïén desmielinització (Wekerle 1994).
Així, desconeguem molts dels aspectes patogènics de la EM, però sembla que hi ha un cert consens en considerar al menys dues aspectes, la presència d'un fator genètic (immunogenètic) i l'actuació d'un agent extern.
La Esclerosi Múltiple. Una malaltía vírica.
Fou Pierre Marie al 1884 (Marie 1884), deixeble i hereu de Charcot qui proposa una etiològia vírica per a la EM, però no va ser fins al 1930 que es comuniqués un primer possible agent responsable quan Chevassut (Chevassut 1930) informà que havía aïllat un agent causal la "Spherula Insulares". En 1948 es publicà que l'agent de la EM sería el virus de la rabia atenuat (Margulis 1948), aquesta hipotesi caigué, però des de llavors molts altres organismes han volgut ser relacionats amb la EM, des del virus del Xarampió fins a la molt recent teoria de l'acantonament del virus de la leucoencefalopatía multifocal progressiva, cap agent exògen s'ha pogut aïllar.
Malgrat aço, la teoria d'un agent infecciós te al menys tres punts on recolçarse: l'epidemiològic; el clínic, amb la demostració d'un augment significatiu d'infeccions víriques que precedeixen a una exacerbació i el coneixement de determinades malatíes desmielinitzants semblants en alguns aspectes a la EM.
Des de la epidemiologia les aportacions més interesant les van fer Kurztke y Hyllested (Kurtzke 1979) on demostraren la presència d'una "epidemia" de EM a les Illes Faroe, després hi han hagut altres estudis assenyalant possibles epidèmies de EM, aquest concepte ha estat revissat (Poser 1988), tot i possant en dubte la seva validessa. Altra dada aportada des de la epidemiològia i que apuntava cap a un agent exògen era l'efecte de les migracions, i així es clàssic considerar que una persona que emigra des d'una país de bais risc a un de d'alt, tot i que té més risc de tenir la malaltía que els seus compatriotes que no han emigrat, aquest risc és menor que el dels nadius del país on emigra (Acheson 1985), aquest fet apunta a que algun factor exògen adquirit durant la infantessa o la pubertat predispossaría al desenvolupament de la malaltía, un últim factor epidemiològic a favor de la presència d'un factor exògen sería la clàssica distribució de la prevalència en funció de la latitut geogràfica de forma que quan augmenta ésta augmenta la EM.
Sense entrar en la evolució natural de la EM hi ha almenys un aspecte en la història natural que fa possible la intervenció d'un agent exògen en la EM, s’ha demostrat qu un 27% de les exacerbacions estaven precedides per una infecció viral (Sibley 1985). Altrament existeixen diverses comunicacions del desenvolupament de EM després d'una infecció per un virus, més concretament després de patir xarampió (015) i també durant una primera exacerbació es va aïllar un virus de l'Herpes Simple (Bergtrom 1989).
En quant als estudis virològics es coneixen diversos agents vírics que produeixen malalties neurológiques caracteritzades per la presència de desmielinització com ara la Panecefalitis Esclerosant Subaguda (PES) (Agnarsdotir 1977), la infecció pel virus JC (Shan 1990) i més recentment les infeccions causades per retrovirus (Greenberg 1995).
D'aquestes tres infeccions la PES està produïda pel virus del xarampió i es caracteritza per lesions desmilinitzants i per la presència de bandes oligoclonals al LCR, en pacients afectes de EM algunes de les BOG reaccionen amb antigens del xarampió (Norrby 1978), la qual cosa podría interpretarse com una infecció persistent per aquest virus, altrament s'ha detectat la presència de RNA de xarampió als cervels de pacients afectes de EM per hibridació "in situ" (Haase 1981). Aquest fets atorguen un possible paper etiopatogènic al virus del xarampió, però els estudis no estan conclossos.
EL virus JC (Berger 1987) és un cas prou especial ja que es tracta d'un virus que infecta especificament l'ologodendrocit i produeix una desmilinització, primer en "plaques" i després confluent,aquest virus és universal i infecta al ser humà durant la infantessa, però sols produeix malaltía en persones immunosuprimides, i a banda de la desmielinització existeix caracteristicament una escassa resposta imflamatòria, la seva relació amb la EM s'ha establert en la observació d'un quadre clínic similar a la EM, anomenat Leucoencefalopatia Remitent-Recurrent asociada al VIH (Berger 1992) , on es va aïllar el VJC al moll de l'òs del pacient juntament amb una infecció del SNC per VIH-1.
Arribem així al possible paper dels Retrovirus en el desenvolupament de la EM, aquesta familia de virus inclou tres grups (Greenberg 1995); els oncornavirus, els lentivirus i els espumavirus, la possibilitat d'induïr una lesió desmielinitzant al SNC molt semblant a la placa d'EM, juntament amb els mecanismes immunopatogènics que fan servir per a desenvolupar la malaltía ha fet que s'investigue profundament els seu paper en la EM (Greenberg 1991, Tendler 1990). Aquesta família de virus es troba ampliament distribuïda en tot el món animal i indueixen alteracions immunologiques que poden donar lloc a una regulació inadecuada del S.I. produïnt alteracions autoimmnues, neoplàsiques o immflamatòries (Greenberg 1995), però per a que aquest procés s'inicïí cal que existisca una "preparació" del organisme receptor de la infecció (Usuku 1988), enllaçant aquesta necessitat amb el segon gran aspecte patogènic, val a dir la susceptibilitat individual a patir EM.
La Esclerosi Múltiple. Malaltía hereditària.
Eichhor al 1896 ja defineix la EM con una malaltía hereditaria i trasmissible (Eichorst 1896), però serà la epidemiologia novament la primera tècnica d'estudi que aportarà dades concretes al voltant d'aquesta especial susceptibilitat a patir EM, així Kurztke ja identifica grups racials amb una incidència i prevalencia molt baixa (Kurtzke 1980) per eixemple els Japonesos (Detels 1977) o els Esquimals , així com la pràctica absència de EM entre els habitants de raça negra de Sudàfrica (Dean 1949). En l’ctualitat estan clarament identificats els grups racials de baix risc (Compston 1999)
Tots els estudis des de llavors duts en païssos d'alta prevalència mostren una baixa incidència i prevalència en aquestos grups racials front als habitants de raça blanca (Compston 1999). Aquests primers estudis ja apunten clarament cap a una susceptibilitata genètica per a patir EM.
En 1972 s'informa per primera vegada la existència d'una relació entre alèls del sistema d'antígens leucocítics humans (HLA) i la EM, obtenintse així una prova directa de la contribució genètica al desenvolupament de la EM (Jersild 1972), malgrat els diferents estudis que relacionant un determinat HLA amb l'augment o disminució de sensibilitat per a patir la EM (Beall 1993), no s'ha pogut demostrat fins ara cap component del sistema major d'histocompatibilitat humà (MHC) necessari i suficient per a produïr la EM. Igualment els estudis de families (Roth 1994) i bessons mono i dizigòtics (Sadovick 1982 i 1994) han possat de manifest que no pot haver-hi una causa exclusivament exògena, així en el primer càs, s'ha demostrat un augment de fins un 25% més alt de patir EM en una dona, germana d'una pacient afecta que en la població general (Sadovick 1988). Igualment, el mètode més clàssic per a estudiar la importància genètica d'una malaltía, aço és, l'estudi de la taxa de concordància entre bessons mono i dizigòtics, dona una tasa de al voltant el 26% per als primers front a un 2,4% dels segóns, qué sería una taxa molt semblant a la taxa de concordància entre germans no bessons (Mumford 1994). Tot i que aquestos resultats poden tenir esbiaixades, sembla clar que d'aquestos estudis hom pot inferir una predisposició genètica a adquirir la EM.
Així doncs, encara que no s'ha pogut aïllar el gen (els gens) resposable de la EM, els estudis bassats en poblacions demostren abastantment l'influència d'aquesta, i a l'hora s'afegit altra dada que possa en dubte la presència d'un factor exògen com és la baixa incidència de EM entre conjugés (Robertson 1997).
La Esclerosi Múltiple. Una malatía d’oritge desconegut.
Vejem com ni la teoría exògena ni la genetísta són capaços d'explicar tot el seguit de manifestacions clíniques o patogèniques, per la qual cosa s'ha postulat i està més o menys acceptat que l'orige de la EM sería una barreja d'ambdòs teories, així es necessitaria la presència d'un agent exògen, el qual actuaría sobre un substrat geneticament propici, donant lloc a un seguit d'alteracions immunològiques que tindríen com a consequència la lesió desmielinitzant i la clínica (De Castro 1994).
Anatomia patològica de la Esclerosi Múltiple.
Des de Charcot (Charcot 1875) la característica específica patològica de la EM és la placa de desmielinització, reblada per Dawson al 1916 (Dawson 1916). Des del principi Charcot va distinguir entre una placa jove i una placa crònica, o placa activa i inactiva, i els estudis s’'han centrat al llarg del segle en definir la distribució de les plaques a les diferents regions i al llarg del SNC (Brownell 1962); la "edat" de la placa, i així diferents autors (Zimmerman 1950, Lumdsen 1951 i Greenfield 1958,) consideraven un estat molt precoç de la placa en la qual no hi haví infiltració celular però si desmielinització, mentre altres autors, (Dawson 1916 i Adams 1952) apuntaven que la hipercelularitat amb augment de la microglia eren els elements inicials de la placa. Posteriorment es considerà que la primera lesió sería la infiltració perivascular limfocitària (Adams 1977).
En qualsevol càs sigua qui sigua la lesió inicial el patró de lesió establerat va quedar fixat a la decada dels 80 pels estudis d'Allen (Allen 1985 i Prineas 1985), des d'aquesta visió la placa temprana establerta quedava formada per una barreja de macròfags i astrocits, desintegració de la mielina, respecte de l'axò i un cert grau d'infiltració limfocitària perivascular mixta de macròfags, limfocits i cèlules plasmàtiques, a mesura que la placa envellíra disminuiría l'infiltrat cèlular imflamatòri i augmentaría la reacció glial, apareguent finalment dany axonal (Raine 1978 i Shintaku 1988). Però la edat de la lesió no diu res de la seva activitat, aquesta vindría donada per l'activitat desmielinitzant al perimetre exterior de la placa, en aquest sentit Prineas sugería que la desmilinització que s'hi produiría al vessant de creixement de la placa sería distint al procés inicial de desmilinització (Prineas 1985).
Si bé als voltants del 60 encara es mantenía que no era possible la remielinització, pronte aquest concepte va caure (Feigin 1966), tot describint-se la possibilitat de la remielinització en la EM, però sobre tot deguem a Prineas, el qual en diversos treballs al llarg de la decada dels 70 va demostrar que aquest fet era possible i constant en la EM (Prineas 1978, 1979 i 1987), fonamental-ment en unes zones de lesion descrites per como a "Shadow plaque", la placa ombribola, (Schlesinger 1909, prineas 1985) però això deixava una pregunta en l'aire: perqué en determinants moments o llocs era possible la remilinització i en d’altres no. A aquesta pregunta s’ha intentat respondrer sobre tot amb els treballs de Lassmann (Luchinetti 1996). al voltant dels distints patrons d'alteració de la Oligodendroglia obrint així la possibilitat a distints mecanismes patogènics que conduirien a una lesió desmielinitzant amb diferents potencials de remielinització.
Fent un repàs dels fets més importants al llarg d'aquest segle, vegem com l'oligodendrocit es situa al centre de tot el procés patologics, nogensmenys és el que forma la vaina de mielina, i tot i que Charcot va demostratr que la conducció nerviosa existía en un axò desprovist de vaina, pronte es va assumir que un axò desprovist de mielina deixava de coduïr i per tant apareixería la clínica (Charcot 1892).
L'oligodendrocit va ser descrit per Robertson (Robertson 1899) i Del Rio Hortega els va donar nom i els caracteritzà (Del Rio Hortega 1921, 1928), al final de la decada dels 70 es va poder calcular, mitjançant tècniques d'autorra-diogràfia, el temps de recambi dels oligodendrocits (Imamoto 1978). Però no ha estat fins la decada dels 90 que ha pogut ser reconeguda la cèlula progenitora del mateix, batejada com a O-2A, una cèlula bipolar que pot donar lloc tant a un oligodendrocit com a un astrocit (Raff 1989). Fins ara doncs, el paper del oligodendrocit sembla central en el desenvolupament de la EM, tant per la seva funció com a productor de la vaina de mielina, com pel fet d'haverse decrit tot un seguit d'anticosso front a components de la mateixa: Proteïna Básica de la Mielina –PBM- (Lisak 1977), glicoproteïna de la mielina-oligodendrocit -MOG- (Linington 1992 –a-), glicoproteïna associada a la mielina –MAG- (Linington 1984), proteïna proteolipídica –PLP- (Sobel 1990), proteïna S-100 beta (Linington 1992 -b-) entre d'altres; i a partir d'aquest moment la investigació es centrà en dos aspectes: el fenomen immnue i la recerca dels gens que podíen codificar els diferents elements de la RI (De Magistris 1992).
Que existía una reacció glial era conegut des del principi, però amb el desenvolupament de la immunopatològia aquesta cèlula, junt amb la microglia han assumit un paper més important, tota vegada qu'els oligodendrocits no expresen HLA del tipus II (Wekerle 1986), i aquestes si (Neumann 1998-a-), per la qual cosa poden actuar en el procés fiosiopatogènic de la imflamació (Neuman 1988-b-).
A mesura que avancem en el coneixement de la patològia de la EM vegem com més estructures del SNC s'impliquen, com ja hem vist s'assumeix el fet que la EM és una malaltía d'orige autoimmne i això fa necesssari que el SNC entre en contacte amb el SI, per la qual cosa la barrea hemato-encefàlia deu de estar damnada, la qual cosa es va demostrar al 64 (Brodman 1964)que la BHE i deprés, aquesta demostració anatomopatológica va ser corroborada per diversos autors a nivel patològic (Adams 1977), en estudis de TAC (Lebow 1978) i sobre tot pels estudis en RM (Kermode 1990), McLean i cols. al 93 inclòs demostraren diferents patrons d'afectació de la BHE en relació amb les formes clíniques de EM (McLean 1993). Arribem amb aquest estudis a la actualitat de la EM on la patología s'uneix amb la immunologia per tal d'explicar la EM, però abans que aço ocurrisca ha hagut al llarg del esgle tot un seguit de definir la malatía i coneixerla clínicament.
Definició clínica de la Esclerosi Múltiple
Ja Charcot en la seva descripció clínica de la malaltía parlà de sospita de EM i de EM possible, arribantse al consens que un únic episodi de disfunció neurológica no podía ser diagnóstic de EM, apareguent un problema que fins ara i encara, no està resolt, es a dir a que anomenen EM?. Hem vist la variabiliatat de dades que la història ens oferix en quant a la anatòmia patològica, la etiològia i la fisiopatògenia, a nivell clínic aquesta heterogeneïtat es trasllada punt per punt. El primer intent de homogeneitzar criteris de diagnòstic de la EM els deguem a Allison i Millar (Allison 1954), que classificaren als pacients en "Disseminated Sclerosis -DS-" temprana, DS probable i DS possible; en el primer càs hom parlava d'una història recent de disfunció neurològica remitent però sense simptomes actuals, la DS probable la definien com una forma renitent-recuurrent sense que hi hagues dubte del seu diagnòstic i la forma DS possible es diagnósticada quan el quadre clínic el sugereix i no hi ha altra explicació, s'introdueix el concepte de progressió.
Les coses están així fins al 1965 quan després de tres anys de treballs es defineixen els criteris de Schumacher (Scumacher 1965), aquests criteris van ser parcialment modificats per Kurztke en 1982 (Kurtzke 1982) (tabla 1). Posteriorment hi hagueren dos intents més de definir criteris diagnóstics per Rose i McDonald i Halliday (Rose 1976, McDonald 1977) (tables 2 i 3), que són variacions dels criteris de Schumacher, i en 1983 es van publicar el criteris de Poser (Poser 1983)
Taula 1: Criteris de Schumacher modificats per Kurtzke (1982)
Anormalitats objectives en l’exàme neurològic, atribuibles a disfunció del SNC
L’examen o la història clínca deuen aportar dades d’afectació en al menys dues o més parts afectes del SNC.
Evidència d’afectació de vies llargues (substància blanca)
Deu d’existir una distribució en el temps:
- Dos o més episodis d’empitloramnet, d’una durada al menys de 24 hores.
- Progressió lenta o a esglaons de signes i símptomes al menys durant 6 mesos.
- Edat a l’inici entre 10 i 50 anys
- Un Metge competent en clínica neurològica deu decidir que la condició del pacient no pot ser atribuida a cap altra causa
(taula 2) que han estat adpotats des de llavors per la comunitat científica tant per al diagnóstic clínic com per als assajos de noves terapéutiques. Aquesta profussió de criteris diagnóstics té la seva explicació en el fet que no hi ha cap marcador biològic per al diagnóstic de la EM i tant la clínica com la evolució natural és extremadament variable.
Taula 2: Criteris de Poser
|
Categoria |
Brots |
Evidència Clínica |
Evidència Paraclínica |
BOG al LCR |
|
A/ E.M. Clinicament Definida |
|
EMCD-A1 |
2 |
2 |
(i) 1 |
|
|
EMCD-A2 |
2 |
1 |
(i) 1 |
|
|
B/ E.M. Definida amb recolçament del laboratori |
|
EMDRL-B1 |
2 |
1 |
(o) 1 |
+ |
|
EMDRL-B2 |
1 |
2 |
(o) 1 |
+ |
|
EMDRL-B3 |
1 |
1 |
(i) 1 |
+ |
|
C/ E.M. Clinicament Probable |
|
EMCP-C1 |
2 |
1 |
|
|
|
EMCP-C2 |
1 |
2 |
|
|
|
EMCP-C3 |
1 |
1 |
(i) 1 |
|
|
D/ E.M. Probable recolçada pel laboratori |
|
EMPRL-D1 |
2 |
|
|
+ |
BOG: Bandes Oligoclonals
LCR: Líquid Cefalorraquídi
Tot i que ja des del principi es va reconeixe un curs crònic i un remitent-recurrent, la observació clínica al llarg dels anys va anar introuïnt subtipus, bassant-se en tres conceptes: la exacerbació-la remissió, la estabilització i la progressió de la malaltía (Weinshenker 1995). El primer problema en definir el curs de la malaltía el trobem en les pròpies definicions dels fenòmens evolutius, així la exacerbació (relapse) que sembla clar que és un agravament de la situació clínica del pacient, va ser definit per McAlpine (McAlpine 1972), segons el criteri de Muller com la aprició d'un nou símptoma o la reaparició d'un preexistent després del primer atac, vam fer la distinció entre excerbació i exacerbació temporal, definida aquesta per l'agreujament d'un símptoma existent amb una duració menor d'uns días, habitualment minuts o hores(Muller 1949). Però va ser Schumacher qui va posar el límit a les 24 horas (Schumacher 1965). Però la definició d'exacerbació encara presentava uns quants problemes, com ara ¿Quan finalitza una exacerbació? i ¿Com finalitza una exacerbació?. McAlpine definí la remissió com la parcial o completa desparició del símptoma (McAlpine 1972), mentre que Confavreux considarà important la distinció entre la excerbació pura, quan hi havía remissió completa i la exacerbació amb seqüela posterior (Confavreux 1980). Així tot i que el concepte d'exacerbació ha quedat clar, no ha esta el mateix amb el concepte de remissió, i encara avui existeixen discrepancies al voltant de la remissió completa o amb seqüel.les i el seu significat pelque fa a la activitat de la malaltía.
Seguint amb el curs de la malaltía, Charcot descrigué "intermissions complètes", (periòdes de remissió), aquests periòdes de remissió junt amb les exacerbacions definirien la forma més clàssica de EM, val a dir la EM Remitent-Recurrent, però com hem vist no tots defineixen la exacerbació igual, així Patzold ho fa com la remissió completa (Patzold 1982), mentre Confavreux com a la estabilització entre exacerbacions (Confavreux 1980). EL temps sembla haver donat la raó a McAlpine i Confavreux, i així avui no es considera la remissió completa necessaria per a la definició de remissió, encara que tampóc hi hagen estudis que determinen quant es considera aquesta i quant dura, igualment quant és el temps d'evolució sostinguda per a considera exacerbació o progressió.
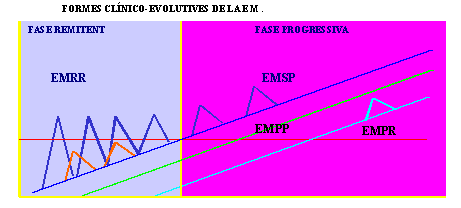 Queda així en suspens la presència d'una fase quiescent o d'inactivitat de la malatia segons el concepte clàsic, que com veurem les noves tècniques s`'han encarregat de desmentir. Amb aquesta dicotomia entre exacerbació-remissió i progressió, entrem en la segona forma evolutiva de la EM, val a dir la EM progressiva. Tot i que la existència d'una fase progressiva de la malatía va ser reconeguda per Charcot , i al llarg del temps s'ha possat en dubte el que fos la mateixa malaltía que la EMRR (Thomson 1997). Les primeres sèries daten de 1949 amb els treballs de Muller, que donava una freqüència del 13% (Muller 1949), des de llavors s'han publicat diferents sèries amb una freqüència de fins el 37% dels casos de EM (Leibowitz 1973), aquesta discrepància assenyala el punt principal de controversia en quant a la evolució de la EM, així Runmarker i Andersen als 10 anys d'evolució d'una cohort de 308 pacients, el 50% tenía una EM progressiva, però en realitat no ho eren de formes primàries (Runmarker 1978), i així cal afegir la tercera forma de EM, val a dir la EM secondaria progressiva (EMSP), que es defineix com una forma de EMRR, però que arrivat un punt inicia una fase progressiva amb o sense exacerbacions, aquesta forma tot i que va ser reconeguda tan pronte com el 1921 (Birley 1921), i que amb posterioritat existeixen diversos estudis que fan referència al moment en que una forma EMRR esdevé EMSP, Patzold i Pocklington no van ser capaços d'identificar el moment en que això ocorriría (Patzold 1982) ha hagut un clar problema de classificació, havet-se utilitzat durant molts anys el terme EM crónica progressiva. Això ha estat així perque aquesta classificació en forma EMSP es sol fer de forma retrospectiva, i no tots els autors consideren igual el concepte de progressió, aquesta divissió és fa més palessa en la aplicació d'aquest concepte per autors estadounidencs, els quals consideren la progressió com el empitjorament continuat durant almenys 6 mesos, siga quin siga la evolució anterior de la malaltía, tot i anomenant a aquesta Esclerosis Multiple Crònica Progressiva (Rao 1988), tot i englovant al menys tres formes clíniques, la EM primaria progressiva, la EMSP i la EM progressiva recurrent, mentre que a Europa sempre s'ha fet una distinció entre les formes de començament a brots i les formes d'evolució progressiva des del principi, tot seguint la classificació de Confavreux (Confavreux 1980). Així vejem com mentre a Europa quden perfilades les formes evolutives en tres tipus als EE.UU no és així, aquesta diferència tidrà importància a l'hora de valorar els assajos clínics i la pròpia patogènia de la malaltía, de fet no és fins al 1996 que s'arriva a un consens en quant a les formes evoltuives (Lublin 1996), a les tres mencionades s'afegeix una quarta la EM progressiva amb excerbacions (EMPE).
Queda així en suspens la presència d'una fase quiescent o d'inactivitat de la malatia segons el concepte clàsic, que com veurem les noves tècniques s`'han encarregat de desmentir. Amb aquesta dicotomia entre exacerbació-remissió i progressió, entrem en la segona forma evolutiva de la EM, val a dir la EM progressiva. Tot i que la existència d'una fase progressiva de la malatía va ser reconeguda per Charcot , i al llarg del temps s'ha possat en dubte el que fos la mateixa malaltía que la EMRR (Thomson 1997). Les primeres sèries daten de 1949 amb els treballs de Muller, que donava una freqüència del 13% (Muller 1949), des de llavors s'han publicat diferents sèries amb una freqüència de fins el 37% dels casos de EM (Leibowitz 1973), aquesta discrepància assenyala el punt principal de controversia en quant a la evolució de la EM, així Runmarker i Andersen als 10 anys d'evolució d'una cohort de 308 pacients, el 50% tenía una EM progressiva, però en realitat no ho eren de formes primàries (Runmarker 1978), i així cal afegir la tercera forma de EM, val a dir la EM secondaria progressiva (EMSP), que es defineix com una forma de EMRR, però que arrivat un punt inicia una fase progressiva amb o sense exacerbacions, aquesta forma tot i que va ser reconeguda tan pronte com el 1921 (Birley 1921), i que amb posterioritat existeixen diversos estudis que fan referència al moment en que una forma EMRR esdevé EMSP, Patzold i Pocklington no van ser capaços d'identificar el moment en que això ocorriría (Patzold 1982) ha hagut un clar problema de classificació, havet-se utilitzat durant molts anys el terme EM crónica progressiva. Això ha estat així perque aquesta classificació en forma EMSP es sol fer de forma retrospectiva, i no tots els autors consideren igual el concepte de progressió, aquesta divissió és fa més palessa en la aplicació d'aquest concepte per autors estadounidencs, els quals consideren la progressió com el empitjorament continuat durant almenys 6 mesos, siga quin siga la evolució anterior de la malaltía, tot i anomenant a aquesta Esclerosis Multiple Crònica Progressiva (Rao 1988), tot i englovant al menys tres formes clíniques, la EM primaria progressiva, la EMSP i la EM progressiva recurrent, mentre que a Europa sempre s'ha fet una distinció entre les formes de començament a brots i les formes d'evolució progressiva des del principi, tot seguint la classificació de Confavreux (Confavreux 1980). Així vejem com mentre a Europa quden perfilades les formes evolutives en tres tipus als EE.UU no és així, aquesta diferència tidrà importància a l'hora de valorar els assajos clínics i la pròpia patogènia de la malaltía, de fet no és fins al 1996 que s'arriva a un consens en quant a les formes evoltuives (Lublin 1996), a les tres mencionades s'afegeix una quarta la EM progressiva amb excerbacions (EMPE).
Però si al cap d'avall s'ha aconseguit definir el curs de malaltía des de la seva patocrònia, més díficl ha estat definir el curs pel que fa al seu pronòstic, i així s'ha intentat una classificació en formes benignes i malignes, fou McAlpine un dels primers en assolir aquest terme, quant després de 10 anys la Escala de disfunció de Kurtzke era igual o menor de 3 (McAlpine 1961), però Poser eleva aquesta definició a 15 anys (Poser 1979), en qualsevol cal admetre que aquest diagnòstic no reflectiría més que un pronòstic, i que al menys en la actualitat cal fer retrospectivament, al no disposar de cap marcado a llarg termini de la discapacitat (Weinshenker 1995).
Així doncs tenim ja la EM definida per criteris clínics, actualment els de Poser, però cal no oblidar que aquests criteris van ser dissenyats per a estudis terapèutics i no serveixen per al diagnòstic de les formes progressives, tenint així que recorrer als criteris de Schumacher per al seu diagnòstic; la podem classificar en una o altra forma evolutiva, però com medim l'impacte d'aquesta malatía en la persona?.
|
Taula 3: Formes Evolutives de la Esclerosi Múltiple |
|
Esclerosi Múltiple Remitent-Recurrent |
|
Esclerosi Múltiple Secundària Progressiva |
|
Esclerosi Múltiple Progressiva amb Exacerbacions |
|
Esclerosi Múltiple Primària Progressiva |
Discapacitat i Esclerosi Múltiple
Aquest repte s'ho va plantejar per primer cop Kurztke al 1955 quan va publicar una escala per a la evaluació de la discapacitat en la Esclerosis Múltiple (Kurtzke 1955), molt semblant a la utilitzada per McAlpine, Compston i Lumsden (McAlpine 1955), aquestes dos escales van estar substituides per la DSS (disability status scale), publicada al 1961 i que evaluava tant el sistemes funcionals afectes com l'impacte d'aquesta afectació sobre la globalitat de l'individu (Kurtzke 1961). Aquesta escla va estar utilitzantse i encara avui està vigent, fins al 1983 quan el mateix Kurtzke publicà la EDSS (expanded disability status scala) (Kurtzke 1983), que introdueix una major sensibiliata en la valoració de la discpacitat. Des de llavors la EDSS ha quedat com la regla d'or per a mesurar l'impacte de la EM en la persona, així com la seva progressió i l'activitat de la malaltía, de fet és el patró que tots els assaigs fan servir com a referència. A més per a una valoració global de la malaltía s'acordà utilitzar una escala mínima d'invalidessa (MRD), que replega la afectació dels sistemes funcionals descrits per Kurtzke, la escala del grau d'invalidessa, la DSS, la EDSS i la ISS (Escala de l'estat d'incapacitat) (IFSS 1984). Tot i que aquesta és la forma actual de mesurar la EM, no és, ni de bon tros perfècta, el que ha fet que hom haja cercat altres scales, entre les moltes utilitzades, cal destacar la SCRIPPS (Sipe 1984), la CAMBS (Mumford 1993), l'index ambulatori -IA- de Hauser (Hauser 1983), i el "Ping Hole Test -PHT-", cada una d'aquestes escales van ser desenvolupades per a guanyar en sensibilitat devant els canvis clínics, que a sovint no replega la EDSS, càs de la SCRIPPS, valorar millor les exacerbacions com fa la CAMBS, la funció dels membres superiors (PHT) o la capacitat per a caminar (IA). Encara i així, queden al menys dos aspectes que en la actualita no es podem mesurar correctament com és l'estat mental i la fatiga, amdòs aspectes tractas grolleramen en la EDSS, i que fins els treballs de Rao (Rao 1991), en el primer càs i de Freal i Murray , però sobretot Krupp, en el segón no han estat prou valorats en la discapacitat i en la definció de progressió i activitat (Freal 1984, Murray 1985, Krupp 1988). Tot aquest esforç te dues explicaciones per un costat la necessitat de dornar un pronòstic i per altra valorar l'efecte dels tractaments emprats, tota vegada que no existeis ni un marcador de activitat ni tant sols un consens al voltant de si hi ha o no activitat clínica.
Tractament de la Esclerosi Múltiple
Malgrat el desconeixement de la etiològia i la etiopatogènia i portats de la mà dels estudis patològics on es veia una infiltració limfocitària junt amb els estudis de LCR amb la demostració de producció intratecal d'immunoglobulines, els primers intents de tractament ja van ser amb immunosupresors/antiinflamatòris.
A l'hora de considera el tractament de la EM hem de fixar ben bé que és allò que volem tractar, així existeixen tractaments purament simtomàtics, com ara el tractament de la espasticitat, tractament dirigits a acurtar el brot, a previndrer la aparició dels brots o a disminuir la progressió de la malaltía i més recentment a intentar la remielinització de la placa.
Miller inicià el tratcament de la Encefalomielitis Aguda Disseminada amb ACTH i posteriorment va fer extensiu aquest ús als brots de EM (Miller 1953 a i b), però la demostració de la efectivitat de l'ACTH va venir d'un assaig aleatoritzat, a doble ceg i controlat amb placebo, el qual va demostrar la efectivitat d'aquesta droga en els brots (Rose 1970), des de llavors la ACTH s'ha vingut utilitzant amb més o menys intensitat, però el desconeixement de la dosi utilitzada i els efectes secondaris, així com la demostració de la absència d'efecte a llarg termini (Rawson 1966 i 1969) va fer que aquest tractament fora substituit pels esteroïds de sintesi (prednisona i metil-prednisolona), el qual va ser iniciat per Dowling a dosis de 2 gr. a 2 1/2 gr día durant dos díes i seguit d'una pauta oral de reducció (Dowling 1980). Variants d'aquesta situació d'han produït, fins a arrivarse a utilitzar pautes gairebé personals. Així en la darrera edició de McALpine's Multiple Sclerosis (Mattews 1991) s'afirma que no hi ha dubte que un curs curt d'altes dosis de metylprednisolona és el tractament d'elecció del brot, però queda oberta la dosi, el numero de díes o la necessitat de continuar les dosis intravenoses amb una pauta de reducció oral. Aquesta línea de tractament assolí la seva màxima importància després de l'estudi per al tractament de la Neuritis Óptica, on a banda de demostrar una recuperació més rapida de la NO, demostrà que en aquells pacients que desarrollaven o ja patien EM, la aparició d'un nou brot trigà més que en el grup placebo, aquest fet introduí un concepte fins llavors del tot insospitat, que era la regulació a llarg termini del Sistema Immune per un tractament curt en el temps (Beck 1993). Aquest concepte va ser représ al 1994 per Kupersmith i cols. que sintetitzaren els possibles mecanismes immunoreguladors del esteroïds (Kupersmith 1994).
Però malagrat la utilització dels esteroïds en dosis altes, encara hi havía un cert numero de brots que no responien a aquest esquema terapèutic, la qual cosa va donar lloc a assajar noves teràpies. Sols altre tractament s'ha fet servir seguint l'esquema d'assaig aleatoritzat, doble ceg i controlad amb placebo, per als brots; el recambi plasmàtic -PE- (plasmafèresi), els dos braços de tractament reberen ACTH intramuscular i ciclofosfamida oral, la conclusió va ser que aquest esquema de tractament era més efectiu per a assolir la remissió del brot, però no tenía efectes a llarg termini (Weiner 1989).
Una vegada comprovat l'efecte dels esteroïds en el brot es va intentar utilitzar aquest tractament per a retardar la progressió de la malaltía, sense que cap esequema terapèutic en base a esteroïds haja demostrat cap efecte sobre la progressió (Fog 1964, Millar 1967, Rinne 1968, Milligan 1987), aquesta constatació va fer que s'escomençes a provar altres alternatives en les formes progressives, referire açi sota l'epigràf progressives sense fer distincions entre la EMSP i la EMPP, car la majoría dels estudis fan referència a la EM crònica progressiva, que com hem vist abans ha estat un terme abandonat.
La primera droga immnusupresora utilitzada per al tractament de la EM progressiva va ser la ciclofosfamida (Aimard 1966, Girard 1967), i encara que els resultats van ser mitjanament bons, la toxicitat i la incomoditat del tractament, que es donava en polsos intravenosos durant 6 setmanes, va fer que es canviés a la Azatioprina (Tucker 1969). Així des de les experiències de Lyon, la ciclofosfamida ha estat un tractament assajat en diverses ocasions i règims (Myers 1987, Killian 1988, Carter 1988), fins al 91 en que el Grup Coopertiu Canadenc publicà els resultats d'un estudi amb ciclofosfamida i PE per a la EM progresiva on no es confirma la estabilització o milloría de la malaltía (The Canadian Cooperative MSSG 1991), a hores d'ara la ciclofosfamida té un interés si més no històric, tot i la relevància que va tenir sobre tot pel fet d'introduïr el concepte d'immunosupressió masiva en el tratament de la EM (Hauser 1983).
Tota una altra cosa és la Azatioprina, inicialment emprada per Tucker amb resultats de dificil interpretació (Tucker 1969), però Silberberg va presentar un assaig ben plantejat malgrat que barrejava formes progressives amb remitents (Silberberg 1973), al igual qu'el disseny de Mertens i Aimard (Mertens 1977, Aimard 1978), la manca d'un disseny adeqüat ha estat el principal problema en la valoració d'aquest fàrmac, així hem d'esperar al 1989, quan Ellison pública el primer assaig doble-ceg, aleatoritzat controlad amb placebo, però encara i així fa servir tres braços de tractament, no arrivant a conclusion clares (Ellison 1989), malgrat aço Hughes al 1992 despés de fer una metanàlisi arriva a la conclussió que donada la bona tolerància i la comoditat del tractament, pot ser d'utilitat en un grup reduït de pacients afectes de EM progressiva (Hughes 1992).
Podríem conclure açi la revissió dels tractaments de les formes progressives de la EM, però els intents de tractament han estat més punyents i intenssos, així la PE va ser assajada amb èxit al 1985 i confirmat el mateix al 91 (Khatri 1985 i 1991), però des de llavors no hi han més experiències amb aquest etractament, tot i quedant com un tractament experimental, possiblement per la perduda de patients al llarg dels anys, amb la perduda de poder estadístic. També s’assajà la ciclosporina sense èxit (Kappos 1988), i tota una sèrie de moderns immnusupressor com la Cladribina (Sipe 1994), el Linomide (Karussis 1996) o la Mitoxantrona (Mauch 1992), de tots aquestos unicament el Metotrexato ha mostrat una certa eficacia en un grup seleccionat de pacients (Goodkin 1995), però encara avui el tractament de la EM progrèssiva romàn una incogníta.
Poser al 1987, va introduïr un concepte de tractament en la EM, que ha estat sens dubte, el més desenvolupat als darrers anys, al seu treball indicava que el restabliment de la funció de la BHE mitjançant l'ús d'esteroïds podría previndrer l'aparició de nous brots i aturar o al menys retardar la progressió de la malaltía (Poser 1987). Si bé aquesta teoria no va reeixir amb els esteroïds, va donar peu al disseny de les noves estratègies terapèutiques tant amb Interferons (The IFNB-MSG 1993), Glatiramer acetat (Jhonson 1995) o Immunoglbulines intravenoses (Achiron 1988), i que han estat fins el moment actual les úniques estratègies efectives en el tractament a llarg termini de la EM.
Així des de l'inici del tractament amb ACTH veiem com han anat evolucionant els assajos clínics, la necessitat de definicions precisses i s'ha fet palés contar amb un mitjà que permitesca valorar l'activitat de la malaltía a curt termini.
Mitjans auxiliars en el diagnòstic de la Esclerosi Múltiple
Entre les diverses tècniques paraclíniques que s'han emprat al llarg de la història contem; amb l'estudi del LCR, i en 1968 quedà establert el valor de l'estudi dels potencials evocats per a demostrar la disseminació de la malaltía, i de fet es consideren en els criteris diagnòstics de Poser (Baker 1968), però la seva validessa com a marcador de progressió de la EM no s'ha pogut demostrar al menys a curt termini, mostrant unicament els potencials somatosensorials del nervi peronial una correlació amb la EDSS i el temps d'evolució (Castilla 1994). Als principis dels 70 es va introduïn la Tomografía Axial Computeritzada (TAC) que permitía veurer per primera vega l'interior del crani, però va demostrat molt poca sensibilitat en casos de EM. La veritable revolució tant en el diagnòstic, el coneixement de la clínica i en el desenvolupament d'estratègies terapèutiques va ser la introducció de la Resonància Magnètica, aquesta teconòlgia és va ser desenvolupada per Bloch i Purcell, rebent el premi Nobel al 1952 per les seves aportacions, al 1977 Lauterbur presentà les primeres imatges (Capdevila 1994) i a partir del 80 es generalitza el seu ús, apareguent el primer article al voltatt de RM i EM en Lancet al 1981 publicat per Young i cols (Young 1981). Des d'un punt de vista històric interesa ressenyar la importància de quatre aspectes: l'aproximació a la patològia "in vivo", l'estudi de la patogènia de la malatía de la EM, l'augment en la sensibilitat diagnòstica i l'intent de convertir aquesta tècnica en un marcador d'activitat de la malatía (Wallance 1992).
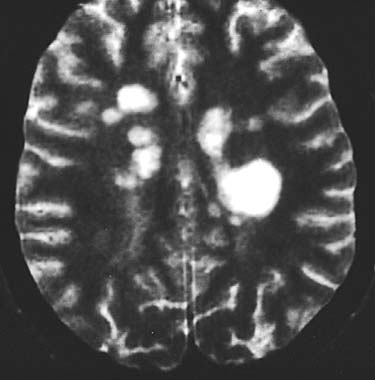 Característicament les images que dona la RM i que estan relacionades amb la EM són les lesions hiperintenses que apareixen en les images potenciades en T2 (Figura 1), i que són el resultat de la substitució de la mielina per aigua, tal i com van descriurer Stewart i Nagara (Steward 1986, Nagara 1987)), però Awad aprecià lesions similars en pacients majors degudes a arterioesclerosi fonamentalment (Award 1986). Newcombe va ser el que estudià més extensament la relació RM-patològia i descrigue tres patrons diferents, arrivant a la conlussió que hi havía una pobra relació entre les lesions hiperintenses en T2 i els canvis atribuibles a plaques de desmilinització (Newcombe 1991). Així en la actualitat es considera que aquestes lesiones en EM poden representar al menys tres tipus diferents de lesions: lesions desmielinitzants, edema, i imflamació (Grossman 1988) (figura 2). Aquesta inespecificitat lesional en T2 tidrà posteriors conseqüències en el disseny d'estudis. Grossmann va ser el primer que va establir una clara relació anatòmica entre els estudis de RM utilitzant gadolinium-DTPA (Gd-DTPA) i la clínica, manifestada aquesta per un nou brot, la significació patològica d'aquesta troballa és la ruptura de la BHE, i tot i que l'aparició d'un nou brot s'ha correlacionat amb la aprició d'una nova lesió en T2, aquesta correlació és més pobra que amb els estudis amb Gd-DTPA, tot i quedant establert que una lesió captant és la expressió d'un brot o d'activitat de la malatía (Smith 1993)(figura 3). Per últim podem apreciar altra lesió, que tot i ser coneguta des del principi sols se li ha donat importància als darrers anys, aço és la lesió hipointensa en T1 i que de forma global va ser definida com una lesió gliòtica (placa cremada) (Barnes 1986) (figura 4), altres autors han donat espècial importància a aquesta lesió, com veurem (Uhlenbrock 1989).
Característicament les images que dona la RM i que estan relacionades amb la EM són les lesions hiperintenses que apareixen en les images potenciades en T2 (Figura 1), i que són el resultat de la substitució de la mielina per aigua, tal i com van descriurer Stewart i Nagara (Steward 1986, Nagara 1987)), però Awad aprecià lesions similars en pacients majors degudes a arterioesclerosi fonamentalment (Award 1986). Newcombe va ser el que estudià més extensament la relació RM-patològia i descrigue tres patrons diferents, arrivant a la conlussió que hi havía una pobra relació entre les lesions hiperintenses en T2 i els canvis atribuibles a plaques de desmilinització (Newcombe 1991). Així en la actualitat es considera que aquestes lesiones en EM poden representar al menys tres tipus diferents de lesions: lesions desmielinitzants, edema, i imflamació (Grossman 1988) (figura 2). Aquesta inespecificitat lesional en T2 tidrà posteriors conseqüències en el disseny d'estudis. Grossmann va ser el primer que va establir una clara relació anatòmica entre els estudis de RM utilitzant gadolinium-DTPA (Gd-DTPA) i la clínica, manifestada aquesta per un nou brot, la significació patològica d'aquesta troballa és la ruptura de la BHE, i tot i que l'aparició d'un nou brot s'ha correlacionat amb la aprició d'una nova lesió en T2, aquesta correlació és més pobra que amb els estudis amb Gd-DTPA, tot i quedant establert que una lesió captant és la expressió d'un brot o d'activitat de la malatía (Smith 1993)(figura 3). Per últim podem apreciar altra lesió, que tot i ser coneguta des del principi sols se li ha donat importància als darrers anys, aço és la lesió hipointensa en T1 i que de forma global va ser definida com una lesió gliòtica (placa cremada) (Barnes 1986) (figura 4), altres autors han donat espècial importància a aquesta lesió, com veurem (Uhlenbrock 1989).
Pel que fa a les aportacions patogèniques, es clar avui día que les lesions que s'iluminen amb Gd-DTPA corresponen a lesions de la BHE, i que aquest fet es pot apreciar tant en fase de brot com en fase de remissió clínica de la activitat, aquesta activitat no detectable clinicament és la major aportació al coneixement de la malatía que ha fet la RM (Stone 1995, Koudriavstseva 1997), també ha servit per a valorar determinades funcions de fàrmacs com l'Inteferon Beta (Stone 1995) i les megadosis d'esteroïds (Barkoff 1991).
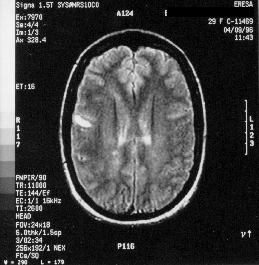 Les aportacions al coneixement clínic són obvies, tant pel que fa a la possibilitat de correlacionar la clínica d'un brot amb una lesió, com pel que fa a la pròpia clssificació evolutiva de la EM, així Koopmans estudia les característiques de la EMPP, trobant diferències significative entre la imatge d'aquesta amb relació amb la EMRR (Koopmans 1989), i ja en el 95 Filippi i cols. estudien els diferents tipus de formes progressives en la EM (Filippi 1995 -a-). També ha servit per a definir una forma de EM, la EM transicional que tot i que encara no està admessa com a forma diferènciada de la EMSP, és possible que un futur aparega com a forma independent (Filippi 1995 –b-). A banda d'aquesta aportació en la història natural, la RM permet diagnòsticar la EM amb un sols brot, i així s'han publicar criteris per a augmentar la especifitat diagnòstica d'aquesta tècnica (Award 1986), inclós Paty va sugerir afegir una subcategoría als criteris de Poser, anomenada EM definidida amb recolzament de RM (Paty 1988), fet que finalment no va ser aceptat. Però el fet és que amb l'adveniment de la RM s'ha utilitzat per a detectar casos subclínics o en familiars de pacients (Lynch 1990). En definitiva la RM ens ha permes augmentar la nostra capacitat diagnóstica i coneixer millor la malatía, però no cal oblidar que és una tècnica molt sensible però molt poc específica com va demostrar Fazekas (Fazekas 1988), així com tots els estudis seriats que s'han realitzat posteriorment, on es demostra la aparició de numeroses lesions noves tant en T2 com amb la RM amb Gd-DTPA (Isaac 1988, Lai 1996), i l'augment en el numero de lesions segons es canvia la tècnia d'obtenció de imatge o la dosi de Gd-DTPA (Filippi 1996 –a-) i el temps de replegada d'imatge (Filippi 1996 –b-, Silver 1997).
Les aportacions al coneixement clínic són obvies, tant pel que fa a la possibilitat de correlacionar la clínica d'un brot amb una lesió, com pel que fa a la pròpia clssificació evolutiva de la EM, així Koopmans estudia les característiques de la EMPP, trobant diferències significative entre la imatge d'aquesta amb relació amb la EMRR (Koopmans 1989), i ja en el 95 Filippi i cols. estudien els diferents tipus de formes progressives en la EM (Filippi 1995 -a-). També ha servit per a definir una forma de EM, la EM transicional que tot i que encara no està admessa com a forma diferènciada de la EMSP, és possible que un futur aparega com a forma independent (Filippi 1995 –b-). A banda d'aquesta aportació en la història natural, la RM permet diagnòsticar la EM amb un sols brot, i així s'han publicar criteris per a augmentar la especifitat diagnòstica d'aquesta tècnica (Award 1986), inclós Paty va sugerir afegir una subcategoría als criteris de Poser, anomenada EM definidida amb recolzament de RM (Paty 1988), fet que finalment no va ser aceptat. Però el fet és que amb l'adveniment de la RM s'ha utilitzat per a detectar casos subclínics o en familiars de pacients (Lynch 1990). En definitiva la RM ens ha permes augmentar la nostra capacitat diagnóstica i coneixer millor la malatía, però no cal oblidar que és una tècnica molt sensible però molt poc específica com va demostrar Fazekas (Fazekas 1988), així com tots els estudis seriats que s'han realitzat posteriorment, on es demostra la aparició de numeroses lesions noves tant en T2 com amb la RM amb Gd-DTPA (Isaac 1988, Lai 1996), i l'augment en el numero de lesions segons es canvia la tècnia d'obtenció de imatge o la dosi de Gd-DTPA (Filippi 1996 –a-) i el temps de replegada d'imatge (Filippi 1996 –b-, Silver 1997).
L’últim apartat que enunciavem, es a dir la recerca de marcador d'activitat de malaltía, s'ha vist, al menys parcialment fustrat, i així encara que existeixen en els diferent assaigs una clara disminució de la activitat mesurada pel volumn lesional total en T2 o als estudis amb Gd-DTPA (Gasperini 1988, Paty 1993, Jhonson 1995), i inclòs ha servit per a finalitzar un assaig abans per la mala evolució de la neuroimatge (Sinnige 1995); la realitat és que no s'ha pogut demostrat una correlació entre aquestes mesures i la progressió de la malaltía (Filipp1 1995 –c-, Stone 1995, Mammi 1996).
Aquest fet ha portat als investigadors a la perpexlitat (Miller 1998), i a hores d'ara està en marxa tota una nova visió de la malaltía, però abans d'entrar en aquest revisaré quins són els fets que han portat a aquesta situació, es a dir els mecanismes fisiopatogènics que al llarg de la història s'han anat propossant.