Hohlfeld en 1997 proposa, després d'una extensa revissió, un "escenari" fisiopatogènic bassat en experiències amb models animals de EAE i observacions en humans, aquest autor replega el sentir majoritari d'aquells que es dediquen a l'estudi de la EM, i així inicia la seva aportació dient que "limfocits T autreactius específics per a la PBM o altres autoantigens del SNC, havent escapat al control tímic de delecció clonal, estaríen en la base del procés autoimmune responsable de la E.M." (Hohlfeld 1997).
Aquesta teoria, que no deixa de ser una hipotesi, es reforça amb altra hipotesi: l'activació de cèlules T ignorants ("naive"), suprimides o anèrgiques a la periferia, fora, del SNC. Com és activat aquest hipotètic clon és un fet desconegut, seguint amb les hipotesis, una tercera sería que aquesta activació inicial ocorriría via "molecular mimicry"-imitació molecular-; en aquest sentit s'ha preparat un repertori de 129 pèptides derivats de virus i bactèris, del qüals set d'oritge víric i ún de bacterià poden activar els limfocits T autoreactius (Wucherpfenning 1995). Altra forma d'activar aquest limfocíts a la perifèria, sería la existència de superantígens que estimularíen als limfocíts T autoreactius (LTA), aquestos LTA seríen activats per encreuament (cross-linking) del seu receptor de cèlula T (RCT) amb una mol.lècula del sistema MHC de classe II expressada en altra cèlula, aquest fet que està demostrat que ocorreix en humans (Marrack 1990, Kotzin 1993), sols exiteixs una evidència indirecta, en estudis amb EAE, del seu possible paper (Brocke 1993). Malgrat aço acte seguit el mateix autor desautoritza aquesta teroria, no ja en EM sino en la mateixa EAE (Brocke1994). Aquests són dos expemples que s'han investigat de la possible activació de l'hipotètic clon de LTA, hi han més, l'autor conclueix que es desconeix el mecanisme d'activació i reactivació posterior del LTA.
Tot seguit i ja donat per fet que el LTA ha estat activat, aquest te que entrar al SNC, Wekerle demostra que la BHE és impermeable a grans mol.lècules i a cèlules, però no a Limfocíts T activats (Wekerle 1986). A partir d'aquest moment la fenomenològia patogènica està ben descrita tant en EAE com en persones afectes de E.M, havent-se descrit els mecanismes mitjançant els quals el limfocít entra al SNC, com el LTA, una vegada al SNC interacciona amb un autoantigen, com es produeix la cascada immflamatòria i per últim la desmielinització i la remielinització, però tots els autors condultats destaquen que aquest és un procés inespecífic.
Arribats a aquest punt començen altra vegada les hipòtesis, així fins ara hem vist que tot el procés està fonamentat en el LTA (hipotètic), en una resposta cèl.lular i en una autoentígen, la PBM, però ocorreix que quant estudiem aquest model no es produeix desmielinització, sent necessari que s'introduixca un anticos monoclonal específic anti MOG, per a que hi hasca desmielinització (Linington 1988, Genain 1995).
Així s'arriba a la conclussió que calen al menys dos fenòmens: A) Imflammació iniciada per cèlules T autorreactives front a determinats antigens del SNC i B) Desmilinització mediada per autoanticossos front a antigens de la mielina o oligodendrocit (Lassmann 1995, Chiang 1996), adquireix així relevància la resposta immune humoral produïda per limfocíts B, i que recordem, és fins ara l'únic element patogènic clarament demostrat en relació a la EM, amb la producció de Bandes Oligoclonas de IgG (BOG). Però el primer element, val a dir la imflammació ¿és realment necessari?, els estudis de Chiang i cols. (Chiang 1996) han vingut a demostrar que la desmielinització induïda per magròfags i microglia pot ocorrer en absència de anticossos específcis front a la mielina o de la resposta celular, i així la producció crònica i mantessa de baixos nivells de IL-3 promou el reclutament, proliferació i activació del macròfag i la microglia produïnt desmilinització primària de la substància blanca del SNC.
Hem repassat les controvèrsies al voltant de la patogènia de la E.M., però cal que ordenem els aconteixements per a millor entendrer que és el que passa a la EM, en qualsevol càs no ens eixirem de la hipòtesi exposada, tota vegada que és la admesa, i en aquesta línia podem classificar els events de la següent manera:
1- Persistència del clons de cèlules T autoreactius.
A- Mecanismes general de la RI.
B- Tolerància immune.
C- Característiques moleculars del L.T. auto-rreactiu.
2- Migració del LTA a traves de la BHE.
3- Reconeixement de l'autoantigen al SNC.
A- Mecanisme de reconeixement de l'antigen.
B- Cèlules implicades en el procés de reconeixement de l'antigen.
C- Molècules implicades en la reacció autoimmune.
C.1- Citoquines.
C.2. Quimioquines.
4- Establiment d'un espai d'imflammació
A- Reacció front a autoantígens.
B- Destrucció de l'antigen (desmielinització).
B.1- Mecanisme de la desmielinització.
B.2- Cèlules implicades en la desmieli-nització.
B.3- Mecanismes humorals de destrucció de la mielina.
C- Limitació del procés imflammatori.
5- Remielinització.
1- Persistència de Clons de Cèlules T autoreactius.
1.A- Mecanisme general de la resposta immune (R.I.).
De forma general el S.I. té com a missió mantenir la integritat biològica front a agressions externes (infeccions) o internes (neoplàsies), mitjançant un mecanisme de reconeixement específic o inespecífic de l'agent agressor. Per a dur a terme aquesta missió el S.I. disposa de cèlules que reconeixen l'agent agressor de forma específica (limfocíts B i T), donant lloc a una resposta immune antigen-específica i cèlules que eliminen patògens de forma inespecífica (monocits i macròfags).
Així el limfocíts T i B (L.T. i L.B., respectivament) porten a la seva superficie receptors específics que reconeixen antigens determinats, aquestos receptor són Immunoglobulines (Ig) en cas del L.B. i receptors antigènics de cèlula T (RCT) en càs del L.T.
Al llarg del creixement de l'individu es produeixen multitud de clons de L.B. i L.T. que van a reaccionar amb patògens determinats, de forma que qüan un patògen entra al organisme es produeix una selecció del clon que va a produïr la destrucció del patògen. Però per a que es produesca aquesta destrucció del patògen, aquest té que ser presentat de forma determinada, així els L.B. s'uneixen a patògens a l'espai extracelular, neutralitzant el patògen a l'unirse al mateix els anticossos que el L.B. porta a la seua superficie, fent-lo així digerible pel fagocít i activant la casacada del complement.
A diferència d'aquest procés, el L.T. no reconeix Atg solubles, processant unicament Atg ligats a la superficie d'una cèlula, la Cèlula Presentadora de l'Antigen (CPA), i més precisament el L.T. reconeix Atg peptídics de carena curta units al Sistema Major d'Histocompatibilitat (MHC).
Exiteixen dos tipus de molècules MHC, les de classe I i les de classe II. Les primeres integren el grup de molècules HLA (Antígens humans leucítics) dels tipus A, B, i C, s'expresen en totes les cèlules de l'organisme unint-se al citosol de la cèlula a Atg patògens, habitualment virus o bactèris, expressan-se a continuació a la superficie celular i activant als L.T. supressor (CD8+), els qüals destrueixen la cèlula infectada o reconeguda com "malalta" pel S.I.
Existeixen cèlules que en condicions normals, es a dir en condicions de no infecció o proliferació tumoral, expresen a la seva superficíe pèptides pròpis, però són ignorades pel S.I., és a dir el S.I. esdevé TOLERANT, per a eixes cèlules.
El MHC de classe II, a diferència dels de classe I s'expressa primariament en cèlules del S.I. (L.B., Macròfags, i L.T. activats), comprenen el grup de molècules HLA DR, DP i DQ, s'uneixen a pèptids derivats de proteïnes degradades a vesicules intracelulars, aquest pèptids poden venir bé d'una font externa o de bactèris o parasits intracelulars, que han entrat bé per fagocitosi/pinocitosi, bé per endocitosi mediada per receptor. Les molècules MHC II activen als L.T. colaboradors (helpper-CD4+), iniciant-se una cascada d'aconteixements que porta a la destrucció del Atg considerat patògen.
Així vejem que per al reconeixement de l'Atg i el desenvolupament de la esposta
immune mediada per cèlules calen molècules MHC, receptors de Cèlula T, receptors específics CD4 o CD8, però aquest esquema s'ha vist complicat per la demostració recent de la presència d'altres molècules necessaries per a la trasducció del senyal, aquestes molecules poden ser de dos tipos: A)Molècules coestimulants; receptors CD28 i CTLA-4 en la L.T. i els seus homòlegs B7-1 i B7-2 en la C.P.A., i B) Les citoquines i els seus receptors. A més el TCR necessita per a actuar adecuadament la presència d'altre recptor existen a tots el L.T. el CD3. Per últim es precissen les molècules d'adhessió per a estabilitzat tot el sistema (figura 13).
Aquest complicat sistema de reconeixement de l'Atg té com a única finalitat que el S.I. reconega lo propi com a propi i lo extrany com a extrany, de no ser així el mateix S.I. destruiría a l'organisme que déu defensar, i aixó és el que passa en càs de les malaltíes anomenades autoimmunes, com sembla ser la EM.
1.B- Tolerància Immune.
El procés mitjançant el qual el S.I. arriba a ser tolerant amb les estructures del organisme no està del tot dilucitat, així s'han descrit els següents:
A) Delecció clonal, que té lloc al timus i contempla dos aspectes una delecció clonal positiva, mitjançant la qual el timus elimina clons de L.T. que no reconeixen molècules pròpies MHC i per la qual cosa no tenen "utilitat"; i una delecció clonal negativa mitjançant la qual s'eliminen clons de cèlules que reconeixen auto-Atg ligats a un MHC pròpi. Així el procés s'inicia per la entrada d'un L.T. precursor al timus i deu aprendre a reconeixer molècule MHC pròpies, i a continuació deu aprendre a tolerar Atg pròpis presentats per MHC. En el primer càs, és a dir la manca de reconeixement de mol.lecules pròpies dona lloc a la delecció positiva, mentre que la segona a la delecció negativa. Malgrat aquest procés de selecció tímica, Wekerle i cols. (Wekerle 1996) demostra que el S.I. és portador de numeroses cèlules capacitades per reconeixer autoantigens expressats al timus, queda així el dubte si els auto-Atg tímics enboirinen (shaped) el repertori immune o el destrueixen.
Aquelles cèlules autoreactives que no han estat destruïdes al timus són controlades per un mecanisme de tolerància perifèrica.
B)Tolerància perifèrica. Comprén dos possibles mecanismes: La inactivació clonal que consiteix en que un L.T. autorreactiu troba la seva molècula MHC pròpia però no dispossa dels coestimulados per a donar lloc a la resposta immune (Schwarts 1996) i un segón mecanisme sería la supressió activa de L.T. autorreactius per L.T. supressors, aquest darrer mecanis sols ha esta enuciat.
Aquestos mecanismes descrits per al L.T. seríen similars per al L.B. (Hodking 1995). Per últim cal dir que no tots els limfocíts autoreactius deuen ser necessariament controlats, ja que; bé per no poder accedir als seus autoantigens, bé per que aquestos s'expresarse en cantitats molt petites, aquest mecanisme es congut com "ignorància Clonal".
En qualsevol càs si algún o algúns d'aquestos mecanismes fallen es produirà una resposta autoimmune i l'aparició de la malaltía autoimmune.
1.C- Característiques del L.T. autorreactiu activat.
El L.T. autorreactiu pot permaneixer força temps sense dividirse ("naive"), però quant entra en contacte amb el seu Atg comença la proliferació clonal i donarà lloc a uns limfocíts efectors (activats) amb capacitat per a destruïr l'Atg, així el L.T. CD4 es capaç de donar 1000 L.T.A. CD4 activats i el L.T.A. CD8 4000 L.T.A. activats (Bottomly 1988). La proliferació i iferienciació del limfocít "naive" (ingenu) estntrolada per la interlucina-2 (IL-2) i la IL-4, i a la vegada el L.T.A. activat produeix IL-2 i receptors IL-2 (rIL-2).
Els L.T.A. activats o efectors expresen a la seua superficie molècules d'adhessió específiques, proteïnes de memebrana implicades en la interacció celular com el ligand Fas (FasL) o la mol.lècula CD40 i citoquines (CQ) determinades.
Existeixen tres tipus de limfocíts efectors, el CD8 que actua front a antigens que, procedents de patògens, operen al citosol i són presentats per MHC-I. I els CD4 que actuen front a patògens presentats per MHC-II i que poden diferneciarse en Th1 i Th2, els primers activen els macròfags i destrueixen als patògens intracelulars, i els segons activen als L.B. per a produïr anticossos, és per aço que de forma estricta aquestos L.T. CD4+ diferenciats en limfocíts funcionalment Th2 seríen els veritablement colaboradors (Bottomly 1988).
Així el perfíl de secreció de CQ en els Th1 sería: la IL-2, l'Interferon gamma, el Factor de Necrosis Tumoral alfa i beta (TNF-alfa), IL-3 i GM-CSF (factor de creixement de colinies de granulocits). Mentre que els limfocíts colaboradors CD4+, Th2, secretarien IL-4, IL-5, IL-6, IL-10 , IL-13 i TGF-beta (factor de creisement tumora-beta). En quant a la seva funció els Th1 activaríen els macròfags, produïrien linfotoxina (LT), mentre que els Th2 actuarien estimulant el creixement i diferenciació de L.B. per a la producció d'anticossos; la diferenciació cap a un L.B. efector, productor d'anticossos (cèlula plasmàtica) o un L.B. memòria dependeix de IL-10 i de les interaccions a través de CD40.
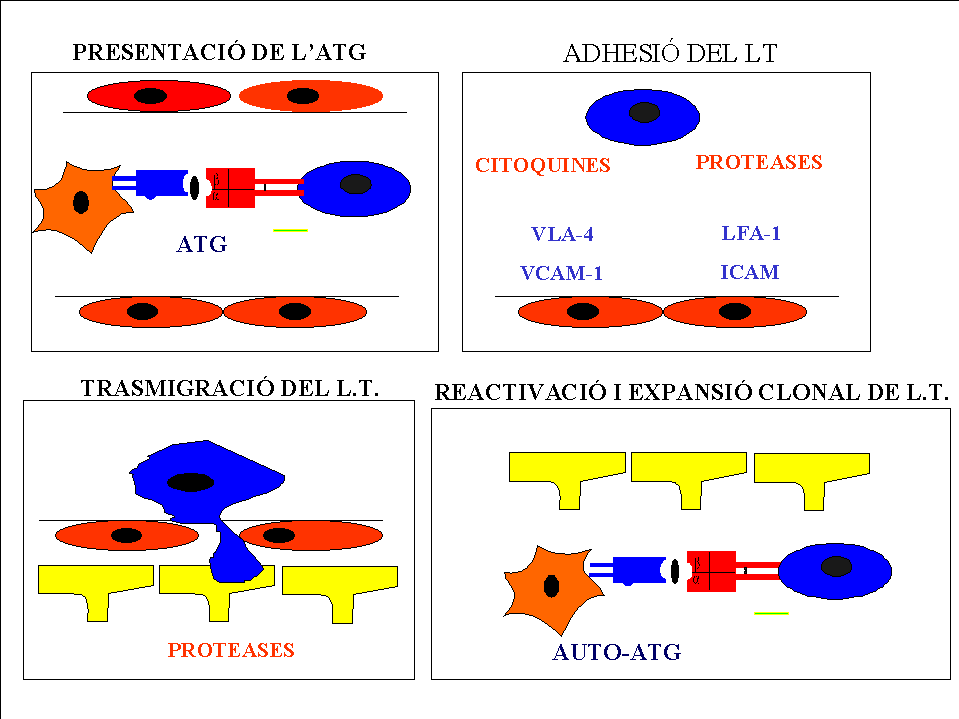
2- Migració a traves de la BHE.
Tota vegada que el limfocít ha estat activat expressa a la seva superficie molecules d'adhessió: Selectines (Slex), l'Antigen-1 Associat al Limfocít (LFA-1) i l'Antigen-4 d'activació molt retardada (VLA-4) (Bevilacqua 1993, Hynes 1992). Aquestes molecules interactuen amb els seus homònims a l'endoteli; selectin, ICAM-1 (molècula-1 d'adhessió intercelular) i VCAM-1 (molècula-1 d'adhessió celular vascular. Finalment el limfocít migra a traves de la BHE seguint senyals derivades del sistema del complement activat (C5a), el factor d'activació plaquetar (PAF) i membres de la familia de les quimioquines (IL-8, IP-10 i NAP-25)(figura 14).
3- Reconeixement de l'autoantígen al SNC.
3.A- Mecanisme de reconeixement de l'antígen.
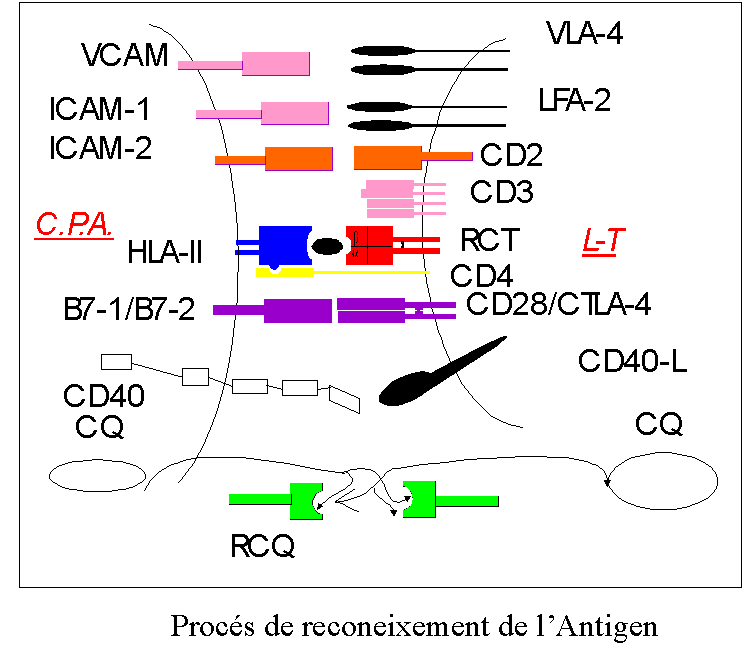
Dues hipotesis són a hores d'ara factibles per a explicar l'inici de la E.M., la primera és la que diu que l'organ, en aquest càs el SNC, és infectat per un virus o altra infecció i les cèlules que infiltren el teixit són reactives front a eixe antigen i a les pròpies cèlules del teixit infectat. L'altra és que l'infiltrat cèlular és autoimune i reconeix proteïnes organ-específiques que són presentats per C.P.A. locals en el contexte MHC apropiat. Si bé qualsevol de les dues hipotesis poden ser certes, el que està demostrat és el fenòmen de difussió de l'epítop ("epitope spreading").
Per a que un autoantigen siga reconegut cal que aproximadament el 0,1% d'una proteïna restritiva MHC de classe II siga ocupada per l'antigen, el que significa que l'antigen déu estar en qüantitas altes (Adorini 1988). En segon lloc un autoantigen front a cèlules T deu ser presentat ligat a una molècula MHC-II d'una C.P.A. al teixit, es a dir, in situ. Al SNC, la microglia pot actuar com a C.P.A., així com l'astrocít, tota vegada que aquest haja estat activat previament, en el context MHC-II.
Inicialment la resposta està focalitzada en epítops immunodominants, però amb posterioritat la cèlula T pot reaccionar front a d'altres epítops "críptics" (Lehmann 1973), perpetuant-se així el fenòmen imflammatòri.
S'ha intentat trobar l'autoantigen que iniciaría el procés imflammatòri al SNC,mitjançant l'activació del L.T. autorreactiu, en aquest sentit s'ha estudiat la PBM i el PLP.
Diversos autors han comprobat per un costat que existeix un numero similar de limfocíts autorreactius front a la PBM i a la PLP en persones sanes i persones afectes de EM, després d'una estimulació antigènica primaria. Però al induir una estimulació amb rIL-2 (recpetor IL-2) les cèlules T reactives eren significativament major en el grup afecte de E.M. que en el control, i al LCR les cèlules T reactives a PBM eren deu vegades més que a les mostres de sang perifèrica, a més aquestes cèlules reconeixien especificament els pèptides 84-102 i 143-168 (Jaraquemada 1990, Ota 1990, Zhang 1990, Martin 1991, Wucherpfenning 1991). En base a aquestes dades es va postular que les cèlules T autoreactives tenen un paper en la patogènia de la malaltía (Hafler 1997).
Per a venir a corroborar aquest fet Lovett-Racke i cols.han demostrat com les cèlules T reactives a la PBM no són bloquejades al eliminar les molèclules coestimulants (Lovett-Racke 1998), la qual cosa significa que aquestes cèlules poden ser activades o diferenciarse en cèlules-T memòria. I tornem açi al fenomen de tolerància, recordem que hem definit l'anèrgia es fruït de la absència de molècules coestimulants, sense les quals no es poden activar les cèlules memòria, aquest treball vé a demostrar que els limfocíts reactius a PBM no depenen d'aquest mecanisme per a ser activats i iniciar la proliferació clonal.
En qualsevol càs aquest estudis vindrien a demostrar la perpetuació d'una resposta immune front a un antigen, resposta que funcionaria com hem descrit al principi, però no dona constestació a quin és l'antigen inicial que actua disparant el procés.
3.B- Cèlules implicades en el reconeixement de l'antigen
.
Una vegada el limfocít T autorreactiu està dins del SNC, dèu confrontarse amb l'antigen, estudis de Wekerle en E.A.E. han demostrat que el limfocìt que penetra és un L.T.CD4+, aquestos L.T. reconeixen antigens presentats per molècules MHC-II (Wekerle 1994), així al SNC les molècules que expresen MHC-II són la microglia, al menys durant els estadis inicials (Fabry 1994), els astrocits, si són activats per CQ proimflammatòries també poden expressar MHC-II (Fontana 1984), i per últim determinades monocits perivasculars (Wekerle 1994, Williams 1994).
Amb el Limfocít T CD4+ activat comença el procés imflammatòri per se, amb la liberació de CQ i l'aplificació de la resposta immune, i pronte altres cèlules locals expresaran MHC-II convertinse així en C.P.A. (Vass 1990, Wekerle 1994, Fabry 1994). El microespai al SNC canvia rapidament amb l'adveniment de noves cèlules del sistema monocit macròfag (S.M.M.) i altres cèlules imflammatòries.
4- Establiment d'un espai d'imflammació
4.A- Citoquines
No hem d'oblidar que la resposta imflammatòria te com a objecte eliminar una substància que es considera nociva, en aquest sentit el que s'estaleix és una resposta equilibrada que destruisca alló que està damnat però no el teixit sà, les CQ seràn les encarregades de mantenir aquest equilibri mitjançant accions activadores i inhibidores de les cèlules efectores de la destrucció tisular.
L'equilibri abans assenyalat es consegueix per la presència de tres tipus de molècules: CQ efectores, CQ inhibidores i molècules inhibidores de CQ. El fi últimés activar les cèlules del S.M.M., les quals mitjançant mediadors d'imflammació donaran lloc a la lesió del teixit diana. Del global de les CQ descrites (Figura 18), descriurem aqueslles en les quals s'ha demostrat un paper en la E.M.
CQ efectores: Es classifiquen en CQ del tipus 1, al estar produides per Th1 i comprenen al TNF alfa i Beta (linfotoxina), La IL-1, IL-2 i l'Interferon-gamma. I CQ del tipus 2, les produïdes per Th2, que integren la IL-4, IL-10, el factor estimulant de colònies de Monocit/macròfag i els Interferons del tipus 1 (Lucas 1995).
Les accions de les CQ proinflammatòries van encaminades totes elles ha produir uan destrucció del teixit, tot i preparant-lo per a que puguen actuar els efector (figura 19) (Hartung 1995).
El TNF-alfa, té un paper primordial en el procés imflammatòri al produïr una regulació a l'alta ("upregulation") de molècules MHC-I i II, molècules d'adhessió, ICAM-1 i VCAM-4, i liberació de productes de resposta inflammatòria com òxid nítric (NO9) i radicals de 02 (Stoll 1993). A més produeix una ihibició dels canals de K a l'oligodendrocit i indueix la seva apotosi (Selmaj 1988). Està produït per limcofíts T, macròfags, microglia i astrocits (Hartung 1995). Actua mitjançant la seva unió a dos receptors transmembrana de 55-70 Kd. S'han identificat receptor soluble per als dos tipus de receptors de TNF-alfa (sTNF-R) (Tracey 1993).
La limfotòxina (TNF-beta), indueix apotosi dels oligodentrocits (Selmaj 1990), mentre que l'interferó gamma té unes accions similars al TNF-alfa (Fierz 1985) La IL-1, de la qual hi han dos tipus; la IL-1alfa i la IL-2beta, que tenen els seus respectius receptors, l'activitat inflammatòria és prouïda pel IL-1R, sent desconeguda l'acció del IL-1 de tipus 2. L'efecte IL-1 es contrarestat per l'antogonista del receptor IL-1 (IL-1Ra), i per receptors solubles de Il-1 tipus 1 (sIL-1R), la situació es força complexa pel que fa a la acció de IL-1, així la activitat de IL-1Ra es veu disminuida per sIL-1R, quant ambdòs s'alliberen junts, però per separat cadasqú antagonitza l'efecte IL-1 (Arebd 1993 i 1994).
Inhibidors de CQ efectores: Actuen impedint la unió de la CQ al seu receptor específic, se han descrit inhibidors de IL-1: IL-1Ra, sIL-1R i sIL-IIR; y de TNF alfa: sTNF-R (Burger 1995).
CQ inhibidores: actuen modulant la producció de CQ efectors i els seus inhibidors, són fonalmentament la IL-4 i la IL-10, aquesta darrear es capaç d'inhibir als linfocíts Th1, mentre que IL-4 produeix una regulació a la baixa ("downregulation") de IL-1 i TNF-alfa.
Amdòs interleuquines disminueixen la expressió de MHC-II pel les cèlules del S.M.M., i una regulació a la baixa de IL-1alfa i IL-1beta, IL-6 i IL-8, TNF-alfa, el factor estimulant de colònies monocít/macròfag i metaloproteases. A la vegada pordueixen una regulació a l'alta de IL-Ra (Burger 1995) (figura 20).
Per últim l'establiment d'un equilibri al lloc d'imflammació intervenen els Interferons, aquestes proteïnes s'han classificat en Interferons del grup 1, que comprén a l'interferon alfa i al beta i Interferons del grup 2, que comprén a l'interferon gamma. Ja hem comentat l'efecte de l'INF-gamma, en quant al paper de l'INF beta, tot i que es una molecula antivírica, el seu efecte immunomodulador bé donat per la regulació a la baixa que fa de l'INF gamma. (Burger 1995).
4.B- Activació del macròfag.
En última instància de tot aquest complicat entramat el que reeix és l'activació del macròfag i la microglia (Perry 1993), aquestes cèlules van a produïr tot un seguit de substàncies: enzimes degradadores de proteïnes (elastases, colagenases, enzimes lisosomials); citoquines i quimioquines: IL-1, TNF-alfa, IL-6, IL-8, IL-12; Radicals lliures; eicosanoïds (prostaciclina, prostaglandina E2, trmboxane A2, leucotriens) i; components del complement. Tot plegat es produirà la lesió del teixit (Hohlfeld 1997).
Tot el procés abans descrit és, si més no el procés inflammatòri, inespecífic, que en teroria dèu portar a la desmielinitazció i a la aparició de la simptomatològia.
4.C- La desmielinització.
La E.M. es distingueix de la resta de mamalties imflammatòries del SNC per la extensió de la desmielinització que acompanya a l'inflitrat imflammatori perivascular (Waubant 1997). Així doncs aquesta desmielinització és o dèu ser important en la E.M. Tres preguntes són importants respondrer en aquest apartat; Com és produeix?, Quan és produeïx?, i Quin significat clínic té?.
En aquest apartat tractaré la primera qüestió, en aquest sentit els estudis assenyalen set possibles mecanismes:
1- Alliberament de CQ, proteases, radicals O2, lipases i altres productes tòxics produïts pel macròfag.
Aquest mecanisme actuaría a traves d'un procés independent de l'antígen i ho faria en la fase aguda i temprana en la evolució de la placa de E.M.(Wisniewski 1975, Sato 1982, Selmaj 1988, Burge 1989, Scolding 1989).
2- Mielinolisis mitjançant l'activació del complement.
Dels mecanismes propossats aquest és el que, al menys en teoria, sembla atansarse més a la realitat, així vejem com el complexe terminal del complement (CTC) puede inserirse a la vaina de mielina i produïr la destrucció de la mateixa formant forats (Hartung 1995).
La font principal del complement és el macròfag, però també la microglia i l'astrocit poden profuïr, al menys algun dels seus components (Levi-Strauss 1987). Existeix evidència d'activació del complement en la EAE, i també en la E.M., mitjançant la detecció de consum de C9 al LCR de pacients afectes de E.M., augment del receptor soluble i els pèptids C3a i C5a al suero i LCR (Sanders 1986, Compston 1986 i 1989). Tambés s'ha demostrat el diposit de CTC a les vaines de mielina (Griffin 1990).
El mecanisme mitjançant el cual es produïria la lesió sería per la abertura de forats que permiterien la entrada de proteases produint la destrucció de la mielina, a més es permetiria la entrada de l'ion Ca++ que donaría lloc a una alteració de la conducció nerviosa (Shin 1988) i la aparició de la simtomatològia.
3- Mielinolisis deguda a la secreció de Ig auto-rreactives, mitjançada per l'activació del macròfag a traves del seu recptor Fc (Waubant 1997).
4- Mitjançant Ig específics dirigits contra components de la mielina (Linington 1988, Lassmann 1988). Aquest mecanisme d'actuar ho faría en fases avançades de la formació de la placa de E.M., ja que diversos autors han estat incapaços de demostrar la presència d'aqueste Ig dirigides contr la mielinia en la placa precoç (Bruck 1994).
5- Inteaccions entre oligodendrocits activats i limfocíts T gamma/delta (Selmaj 1991).
6- Dany directe sobre l'oligodendrocit amb afectació secondaria de la mielina, derivat de la secreció de TNF-alfa i beta per les cèlules Th1+ (Waubant 1997).
7- Lisis de l'oligodendrocit per LT CD8+ i en menor mesura LT CD4+, mediada pel Sistema MHC-I no restringit (Waubant 1997).
5- Finalització del procés inflammatòri.
El procés mitjançant el qual finalitza la inflammació no està del tot ben establert, però alguns dels fenòmens que ocurreixen són:
A) Augment de la producció de citoquines inhibidores: Il-4, IL-10 i TGF-beta (Rieckmann 1994).
B) Apoptosi dels linfocíts autorreactius (Scmied 1993), la mort programada d'aqueste celules pot ser la conseqüència de l'agument de la producció d'esteroïds, l'equilibri entre citoquines, l'absència de disponibilitat de molecules co-estimuladores, o també induït per antigens (Bauer 1995).
C) Augment de les cèlules amb activitat supressora (Hartung 1997).
En qualsevol càs el procés imflammatori termina, unes vegades amb recupreació completa, altres deixant seqUel.les, d'altres progresa de forma rapida (forma Marbug) i sembla que sempre exiteix una imflammació, de baix grau persistent al Sistema Nervios Central. Condicionat les distintes formes evolutives de EM.
Però, aquest espai fiosiopatogènic, té alguna correlació amb el que coneixem de la anatomia de la E.M.? o de la seva evolució clínica?.