![]()
EL SUEÑO EN LAS ENFERMEDADES PRIONICAS
M.A. Merino Ramirez, M. Escudero Torrella
Servicio Neurofisiología Clínica.
Unidad de sueño
Hospital Universitario Dr. Peset.Valencia
E-Mail:
mmerinor@meditex.es
mescuderot@meditex.es
INTRODUCCION
Desde la primera descripcion de la enfermedad de Creutzfeld-Jakob, ( ECJ ) varias formas de encefalopatias esponjiformes han sido descritas:la aparición de otras formas de la enfermedad, como la nueva variente de C-J, la enfermedad de Gerstmann-Sträussler-Scheinker (GSS) y el insomnio familiar fatal (IFF) y formas de gliosis subcortical familiar progresiva, cuya causa comun de todas ellas es la presencia de una proteina celular alterada designada como prion.
Recientemente por los importantes avances en patologia, genetica y la aparición de nuevas técnicas de inmunohistoquimia para la proteina prionica (PrP), ha sido posible conocer casos clínicos de demencias talamicas y gliosis subcortical progresiva, que aunque en principio no se sospechara fueran causadas por priones, presentan sin embargo deposito de proteina prionica. (Collinge y cols), y se han descrito una serie de variantes, algunas bien definidas y otras con limites poco precisos.y donde el polimorfismo genetico parece determinar las diferentes variantes familiares de ECJ y la IFF.
(12)
La afectación importante en estas afecciones de las estructuras talamicas, centros responsables del desarrollo del ciclo normal vigilia sueño supone la aparición de uno de los síntomas clínicos predominantes en estas enfermedades.(35) En este trabajo realizaremos una descripción clínica y neurofisiológica de los hallazgos EEG de sueño.
INSOMNIO FAMILIAR FATAL ( IFF )
El IFF fue descrito por Lugaresi y cols en 1996 (1). Se trata de una enfermedad hereditaria, de evolución muy rápida, caracterizada por la presencia de un insomnio progresivo y rebelde al tratamiento, acompañado de manifestaciones autonomas y somatomotoras (21-23). Se ha encontrado una degeneración casi selectiva de los núcleos talámicos ventral anterior (VA) y dorsomedial (DM); junto a la presencia de deposito de proteína priónica anómala en el cerebro. En todos los casos se ha confirmado la existencia de una mutación puntual, del codón 178 del gen de la proteína priónica (cromosoma 20). Esta consiste en la sustitución del ácido aspártico por la asparagina. Los hallazgos genéticos y anatomopatológicos de esta enfermedad sugieren que las demencias agrupadas hasta ahora bajo el término de "demencias talámicas" pudieran ser en realidad casos de insomnio familiar fatal u otras variantes clínicas relacionadas con las enfermedades por priones (4).
EPIDEMIOLOGIA Y CLINICA
Hasta la fecha, sólo se han identificado 6 casos en Italia ),(5),(3 familias, un caso con historia familiar negativa,10 casos en Francia(6) (3 familias),9 casos muy interesantes en Alemania (8),(8 familias, con historia familiar sugerente sólo en 4 casos), 5 casos en Austria(7), (1 familia), 2 casos en Gran Bretaña(9), (inicialmente diagnosticados como ECJ y con historia familiar poco detallada), 1 caso en Japón(10), (con historia familiar negativa), 4 casos en Australia(10), (2 familias. Uno de los casos es el paciente más joven conocido con esta enfermedad, 20 años, y su debut no incluyó aparentemente el insomnio) y al menos tres familias más en Estados Unidos,(10) con 6 casos documentados como mínimo (5-10).
La presentación clínica es uno de los aspectos que convierten al IFF en una enfermedad heterogénea. A pesar de unas características genotípicas idénticas, la edad de debut y el curso evolutivo son variables (7). En función de la evolución de la enfermedad y su correlación con el polimorfismo del codón 129 se ha aceptado la existencia de 2 variantes principales: la de rápida progresión (D178N,129M) y más larga evolución (D178N, V129M) o forma atípica, fácil de confundir inicialmente con las formas atípicas de ECJ(D178N, M129V) (11).
Aunque la mayoría de los casos de IFF son hereditarios (patrón autosómico dominante), algunos parecen presentarse de forma esporádica (5,8). La edad media de inicio de la enfermedad es de 49 años (20-72 años). La enfermedad progresa irremediablemente hasta la muerte del paciente, con una media de 15 meses desde el inicio del proceso ( 7-37 meses).
El síntoma más frecuente de inicio es el insomnio progresivo, que empeora hasta llegar a una situación de pérdida casi absoluta del sueño. Los trastornos neurovegetativos son también precoces, pudiendo aparecer hipertermia, hipersudoración, miosis, impotencia, elevación de la frecuencia cardiaca y la tensión arterial, alteraciones urinarias y estreñimiento.
Las manifestaciones neurológicas pueden presentarse al inicio o al cabo de unos meses de evolución, y consisten en disartria, ataxia e hiperreflexia. El habla se vuelve ininteligible y la ataxia de la marcha se agrava hasta una situación de astasia-abasia completa. Más tarde, aparecen mioclonias segmentarias o difusas (espontáneas o provocadas) y en fases tardías pueden estar presentes diplopia y alteraciones de los movimientos oculares sacádicos, distonia y crisis generalizadas tónico-clónicas.
Los estudios neuropsicológicas muestran alteraciones de los lóbulos frontales, afectándose especialmente, la vigilancia, atención y las capacidades visuales y/o motrices, junto a déficits de memoria selectivos. Aunque, no se encuentra una afectación global de las capacidades intelectuales, sino más bien una afectación progresiva de las capacidades de atención y vigilancia (4, 26). En la fase terminal el paciente presenta una incapacidad para conciliar el sueño, síndrome confusiónal, agitación motora y episodios alternantes de vigilia y subvigilia estuporosa, en las que el paciente ejecuta movimientos y gestos que parecen relacionarse con el contenido de un sueño. Se llega a una situación de estupor, con pérdida de los ritmos circadianos hormonales e inestabilidad, terminando en un coma irreversible, con importantes fluctuaciones de la tensión arterial e hipotermia.
La función hipotálamo-hipofisaria es normal. Sin embargo se puede observar una pérdida o reducción del ritmo circadiano de la secreción de hormonas tanto ligadas al sueño (GH, PRL) como de otras que no lo están (Cortisol, ACTH). Las concentraciones plasmáticas de catecolaminas y cortisol se mantienen constántemente elevadas por encimas de los límites de normalidad, manteniéndose consecuentemente reducidos los niveles de ACTH. La Melatonina no muestra el característico aumento de su secreción al pasar de la luz a la oscuridad. En 5 casos de IFF descritos en Austria, se presentó una rápida y severa pérdida de peso en todos ellos en fases muy precoces de la enfermedad (7)
NEURORRADIOLOGIA
La pruebas de neuroimagen (TAC y RNM) son normales. Los estudios de Tomografía por Emisión de Positrones (TEP) muestran hipometabolismo de la glucosa en el tálamo (en particular de la porción anterior) , el putamen cerebelo y, en dos casos, las cortezas cerebrales límbica y parietotemporal (25).
GENETICA:
Todos los casos de IFF presentan una mutación puntual del codon 178 (6,17,22 ). Esta mutación ha sido observada en ciertos casos de ECJ familiar (3), incluso en las familias estudiadas por IFF se han observado dos casos de ECJ ( 27 ). Sobre el cromosoma mutado en posición 178, el codón 129 codifica una metionina en las familias de IFF en vez de la valina codificada en las formas familares típicas de ECJ. La duración del IFF es más corta en los paciente homocigóticos MET/MET sobre el codón 129, mientras que la presencia de una VAL en el codón 129 (forma heterocigota) prolonga característicamente la evolución clínica de la enfermedad (Fig. 1).
HALLAZGOS NEUROFISIOLOGICOS
1. ELECTROENCEFALOGRAFIA:
La actividad de base electroencefalográfica se caracteriza inicialmente por ritmos alfa lentos que, progresivamente, se van extendiendo a las regiones cerebrales anteriores. Posteriormente la actividad cerebral se hace más lenta y difusa así como menos reactiva a estímulos (24). En algunos pacientes, en fase terminal, se han podido observar descargas periódicas o pesudoperiódicas a 1-2 Hz, sincronizadas con las mioclonias.
2. POLISOMNOGRAFIA:
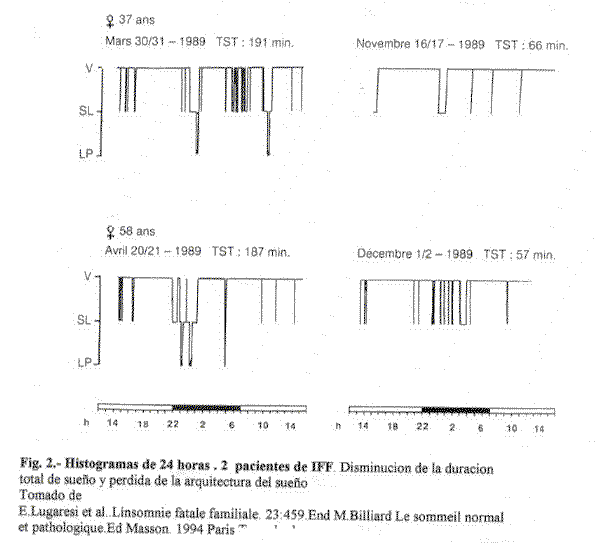
Los estudios poligráficos repetidos ,de 24 horas, realizados por Lugaresi, han mostrado una reducción progresiva de la duración del sueño, hasta quedarse reducido a escasamente unos minutos de duración. La transición de la vigilia al sueño lento y viceversa acaba siendo instantánea, sin transiciones, asociándose a rápidas modificaciones cardiocirculatorias. Inicialmente, en fases muy precoces de la enfermedad, los pacientes muestran un desinterés por su entorno. Incapaces de mantener la atención, responden rápidamente a cualquier estímulo, pero casi con la misma rapidez caen de nuevo en un estado de absoluta falta de atención (42). Los pacientes, dejados completamente solos, tienden a permanecer en un estado de aparente somnolencia, presentando episodios, sin transición, de desincronización y movimientos oculares rápidos que aparentan un sueño REM abortado: episodios de breve duración, atípicos, asociados a abundantes actividades motoras y gestos que evocan aparentemente el contenido de un sueño. El aumento del tono muscular sugiere un sueño REM sin atonía.
En esta etapa, los estudios polisomnográficos muestran una situación de ausencia completa o casi completa de los patrones fisiológicos electroencefalográficos del sueño (fig.2), con mala definición de los estadios. Los husos de sueño (spindles), desaparecen precozmente, y la actividad delta también lo hace posteriormente, reemplazándose por una actividad theta de escasa amplitud a 4 Hz. En definitiva, el paciente llega a una situación en la que existe una pérdida completa del sueño fisiológico, generándose únicamente y de forma muy breve y esporádica episodios de sueño REM o, más infrecuentemente todavía, de sueño delta, surgiendo anómalamente desde la vigilia (41).
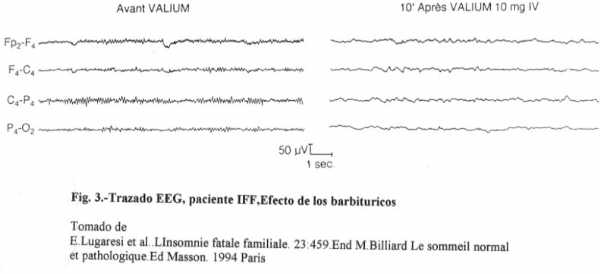
El sueño (o el coma) farmacológicamente inducido por los barbitúricos y las benzodiacepinas se asocia a un aplanamiento de los trazados EEG, no observándose las actividades rápidas o lentas típicamente provocadas por estos fármacos (fig.-3). Como se puede ver, la alteración de sueño que sufren estos pacientes y que caracteriza la enfermedad no puede ser comparada con el insomnio trivial que mucha gente (15-20%) puede presentar a lo largo de su vida.
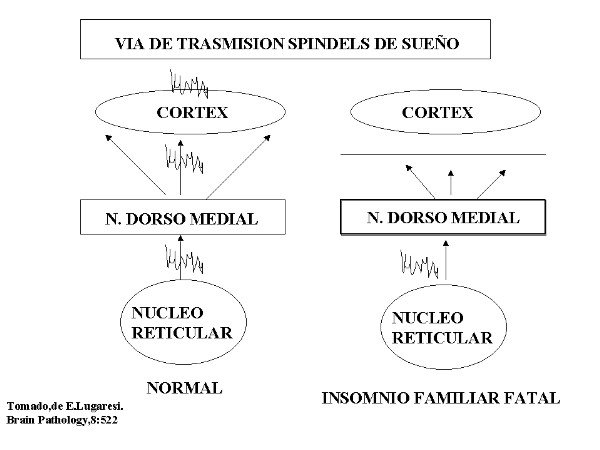
En el IFF, el "insomnio" se refiere a una compleja y permanente situación de la alteración del ciclo vigilia-sueño, caracterizada por la incapacidad para producir un patrón cíclico fisiológico de las actividades típicas electroencefalográficas (EEG) del sueño NREM y REM, así como de las fluctuaciones circadianas hormonales y vegetativas. La transición de la vigilia al sueño parece alterarse de forma significativa, como queda demostrado electroencefalográficamente por la desaparición precoz de los husos de sueño, los cuales juegan un papel primordial en la suave transición de la vigilia al sueño, entre el sueño NREM y REM, y viceversa (Fig. 4).
3. POTENCIALES EVOCADOS:
Los potenciales evocados somatosensoriales, visuales y auditivos de troncocerebral son normales en todos los pacientes estudiados.
4. RESPUESTA PSICOGALVÁNICA SIMPATICOCUTANEA:
La elevación de la temperatura corporal, la hipersudoración, la elevación de los ritmos respiratorios y cardiaco y la hipertensión arterial sistémica son secundarios a una hiperactividad simpática de fondo (con niveles de adrenalina y noradrenalina permanentemente elevados). Los estudios neurofisiológicos sobre la respuesta simpaticocutánea han mostrado unas respuestas disminuídas o ausentes.
DIAGNOSTICO:
El diagnóstico clínico de la enfermedad es relativamente sencillo cuando el paciente tiene una historia familiar de posible enfermedad neurodegenerativa y se manifiesta con trastornos del sueño (insomnio), signos vegetativos y una combinación de signos neurológicos focales. Sin embargo, se han descrito casos de IFF en los que las alteraciones del sueño (insomnio) o bien han estado ausentes durante la fase precoz de la enfermedad o han sido muy leves durante su evolución (8), e incluso en ocasiones el insomnio ha permanecido ausente a lo largo de la enfermedad (10). Además, la aparición precoz de trastornos del sueño, frecuentemente referidos como insomnio, también están presentes en casos esporádicos y familiares de ECJ (10-15 %) (13, 14). En algunos casos de ECJ, el insomnio llega a simular la severidad del IFF (15). En el estudio Alemán de seguimiento epidemiológico de la ECJ, se observaron alteraciones significativas del sueño en casi el 30% de los casos de ECJ esporádica, en algún momento de su evolución (8).
FISIOPATOLOGIA
Los cambios histopatológicos post-mortem y los estudios in vivo mediante PET de los pacientes con IFF, en todos los casos, muestran anomalías en la región talámica y las áreas límbicas corticales relacionadas (43). Es lógico pues, atribuir la clave de las manifestaciones clínicas de la enfermedad (alteraciones del binómio vigilia-sueño y de la función autónoma) a las lesiones encontradas en el tálamo, sin infravalorar la presencia de PrP en el hipotálamo y en el tronco de encéfalo, cuya presencia debe jugar algún papel en la alteración del ciclo vigilia-sueño (44).
En el IFF, la atrofia del núcleo talámico DM podría ser responsable de la interrupción de las conexiones entre los núcleos RE y la corteza prefrontal; conexiones consideradas fundamentales en el establecimiento y mantenimiento del sueño. La propia degeneración de las estructuras talámicas citadas probablemente causan la desconexión entre la corteza límbica y el hipotálamo desencadenándose una pérdida del equilibrio interno viscero-endocrino, con fluctuaciones hormonales circadianas cada vez más importantes y una progresiva y severísima inestabilidad del sistema vegetativo autónomo, con una hiperactividad inicial catecolaminérgica que acaba finalmente en un caos.
Hay que destacar, que los estudios polisomnograficos pueden aportar hallazgos sobre la existencia del insomnio en estos pacientes, cuando la clínica no evidencia este trastorno.
EL SUEÑO EN LA ENFERMEDAD DE CREUTZFELD-JAKOB
Los pacientes con ECJ, presentan frecuentemente trastornos del sueño como manifestación clínica de inicio .Aunque no suele ser el sintoma predominante, en algunos casos pueden comportarse simulando un IFF. En algún trabajo ( 8 ), se citan alteraciones significativas del sueño en casi el 30% de los casos de ECJ esporadica en algun momento de su evolucion.
POLISOMNOGRAFIA
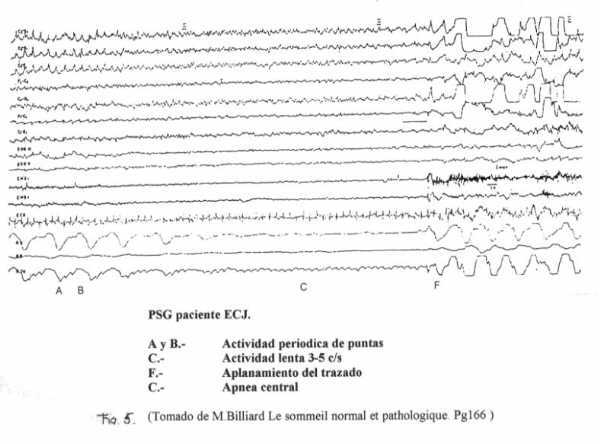
Los pacientes con ECJ sufren precozmente una total desorganización de los ritmos de vigilia-sueño, apreciándose con frecuencia un trazado alternante en el que se suceden las fases de descarga de complejos periódicos generalizados (clasificadas como fases A y B según presenten o no movimientos asociados) y las fases de atenuación del trazado con disminución en la amplitud de la actividad EMG (fase C) y desaparición de las puntas periódicas. Los movimientos oculares pueden estar presentes durante los periodos de la fase C. En la fase de debut de la enfermedad es posible registrar durante la noche una actividad poligráfica que podría corresponder al sueño paradójico o sueño REM (estadios D y E).
El sueño no es fácilmente reconocible clínicamente dada la naturaleza de la enfermedad. Los estudios poligráficos se constituyen en una herramienta imprescindible para su análisis, dándose además la circunstancia de que los diferentes estadios clasificables del sueño no son observados en estos pacientes.
Durante el curso de la noche se puede observar una alternancia de los ciclos A,B y C, con un periodo de duración de 3 o 4 minutos. Esta sucesión se ve interrumpida por los periodos de vigilia, de sueño contínuo y en la fase de debut por las fases que podrían corresponder al sueño paradójico. Las apneas sobrevienen igualmente durante las fases del sueño. (Fig.5)
La afectación precoz de los patrones de sueño sugiere la implicación específica de estructuras involucradas en su génesis. La profunda desestructuración del sueño puede explicarse por la extensión subcortical de las lesiones en esta encefalopatía.
COREA FIBRILAR DE MORVAN
Esta enfermedad esta considerada de forma diferente segun diversos autores. Algunos consideran una variante de las actividad continua de la fibra muscular (ACFM),(29) por exposicion cronica a ciertos toxicos, como el mercurio y las sales de oro, mientras que otros autores consideran probable la causa infecciosa ( particulas viricas o priones ). Es un trastorno extraordinariamente raro cuyos sintomas principales incluyen el insomnio severo,(28) las halucinaciones, fasciculaciones musculares, mialgias eritema y edema en extremidades. Las fasciculaciones a veces importantes, respetan musculatura facial y no cesan con el sueño o la anestesiageneral. Son caracteristicas las descargas neuromiotonicas.
Con el progreso de la enfermedad, hay reduccion importante en la cantidad de sueño. En las fases iniciales de sueño, hay mialgias, halucinaciones y fenomenos de vasocontriccion, sobre todo en piernas. El cuadro evoluciona durante meses hasta el exitus.
Los hipnoticos habituales son ineficaces, solo la administracion masiva de 5 hidroxitriptofano, hasta 12 g/dia, mejora el imsomnio de forma transitoria.
POLISOMNOGRAFIA
Existe una marcada reduccion del tiempo total de sueño. Perdida progresiva de las diferentes fases de sueño quedando solo en etapas finales la fase I. Son frecuentes y caracteristicas las halucinaciones ( visuales, olfatorias y somaticas ), durante los periodos de inicio del sueño.
Bibliografía
- Lugaresi, E., Medori, R., Montagma, P. et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N. Eng. J. Med. 1986; 315(16):997-1003.
- Goldfarb L.V., Petersen R.B., Tabaton M, et al. Fatal Familial Insomnia and Familial Creutzfeldt-Jakob disease: disease phenotype derermined by a DNA polymorphism. Science, 258, 806-808, 1992.
- Lugaresi, E., Montagna, P. Fatal Familial Insomnia: A new prion disease. In: Kryger M.H., Roth, T.,Dement, W.C. Principles and Practice of Sleep Medicine. Saunders WB 1994; 547-548.
- Maurizio, P., Ladogana, A., Petraroli, R, et al. (1998). Recent Italian FFI Cases. Brain Pathol. July, 8:564-566.
- Julien, J., Vital, C., Delisle, MB, et al. (1998). The French FFI Cases. Brain Pathol. July, 8:555-558.
- Budka, H., Almer, G., Hainfeliner, JA, et el. (1998). The Austrian FFI Cases. Brain Pathol. July, 8:554.
- Kretzschmar, H., Giese, A., Zerr, I., et al. (1998). The German FFI Cases. Brain Pathol. July, 8:559-561.
- Will, RG., Campbell, J., Moss, TH., et al. (1998). FFI Cases from the United Kingdom. Brain Pathol. July, 8:562-563.
- Brown, P., Cervenáková, L., Powers, JM. (1998). FFI Cases from the United States, Australia, and Japan. Brain Pathol. July, 8:567-570.
- Sheilhean, D., Duyckaerts, C., Hauw, J,J. Insomnie Fatale Familiale et Maladies à Prions. Rev. Neurol. (Paris), 1995, 151, 4, 225-230.
- Montagna, P. et al. (1998). Clinical features of fatal familial insomnia: phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol. July, 8:515-520..
- Terzano, MG., Parrino, L., Pietrini, V., et al. (1995). Precocious loss of physiological sleep in a case of Creutzfeldt-Jakob disease: a serial polygraphic study. Sleep 18.
- Chapman J, Arlazoroff A, Goldfarb LG., et al. (1996). Fatal Insomnia in a case of familial Creutzfeld-Jakob disease with the codon 200Lys mutation. Neurology 46:758-761.
- Brown P, Gibbs CJ, Rodgers-Johnson P., et al. (1994). Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 35:513-529.
- Petersen RB, Parchi P, Richardson SL., et al. (1996). Effect of the D178N mutation and the codon 129 polymorphism on the metabolism of the prion protein. J. Biol Chem 271, 12661-12668.
- Cortelli P, Parchi P, Contin M., et al: Cardiovascular dysautonomia in fatal familial insomnia. Clin Aautonom Res 1:15-21,1991.
- Medori R, Tritschler J, LeBlanc A, et al: Fatal familial insomnia, a prion disease with a mutation at codon 178 of the prion protein gene. N Engl J Med 326:444-449,1992.
- Lugaresi E: Thalamus and sleep. Neurology 42(Supp 6):28-33,1992.
- Tinuper P, Montagna P, Medori R, et al. Fatal familial insomnia: the thalamus participates in the regulation of the sleep-waking cycle. A clinico-pathological study in fatal familial thalamic degeneration. Electroencephalogr. Clin. Neurophysiol., 73, 117-123. 1989.
- [18 F]FDG PET in fatal familial insomnia: the functional effects of thalamic lesions. Neurology. 1993 Dec; 43(12): 2565-2569.
- Gallassi R., Morreale A., Montagna P., et al. "Fatal Familial Insomnia": Neuropsycological study of a disease with thalamic degeneration. Cortex. 28,175-187,1992.
- Manetto V, Medori R., Cortelli P., et al. Fatal familial insomnia: clinical and pathologic study of five new cases. Neurology, 42,312-319,1992.
- Martin J-J (1994). The thalamus: anatomoclinical correlations. In: Guilleminault C., Lugaresi E.,Montagna P., Gambetti P. (eds). Fatal familial insomnia: Inherited prion diseases, sleep and the thalamus. pp. 45-55. Raven
- Sforza E, Montagna P, Tinuper P., et al. (1995). Sleep-wake cycle abnormalities in fatal familial insomnia. Evidence of the role of the thalamus in sleep regulation. Electroencephalogr Clin Neurophysiol 94: 398-405.
- Gallassi R, Morreale MD, Montagna, et al. (1996). Fatal familial insomnia: behavioral and cognitive features. Neurology 46:935-939.
- Corte, P., Perani, D., Parchi, P., et al. (1997). Cerebral metabolism in fatal familial insomnia: Relation to duration,neuropathology, and distribution of protease-resistent prion protein. Neurology 49:126-133.
- Parchi P, Castellani R, Cortelli P, et al. (1995). Regional distribution of Protease-resistant Prion Protein in Fatal Familial Insomnia. AnnNeurol 38:21-29.
- Wooten, V. Fibrillary Chorea (Morvan´s Chorea). In: Medical causes of Insomnia. Kryger M.H., Roth, T.,Dement, W.C. Principles and Practice of Sleep Medicine. Saunders WB 1994; 547-548.
- Fernandez;J.M;Codina Puig-Grosa;A :inTratadoNeurologia .Ed Aran.1996; cap,56: 911-917