![]()
ELECTRODIAGNÓSTICO EN LOS TRANSTORNOS DE TRANSMISIÓN NEUROMUSCULAR
Chumillas MJ y Cortés V.
Servicio de Neurofisiología Clínica
Hospital Universitario La Fe. Valencia. España.
Todos los procesos que implican un transtorno de la transmisión neuromuscular van a confluir en la expresión clinica común de fatigabilidad desencadenada por el ejercício y condicionada fisiopatológicamente por una alteración pre o postsináptica del factor de seguridad de la placa motora.
La función del electrodiagnóstico consiste en demostrar dicha alteración1 excluyendo o confirmando otras enfermedades neuromusculares coexistentes2,3, apoyando el seguimiento y la monitorización terapéutica de estos procesos 4,5.
BASES FISIOPATOLÓGICAS DE LA TRANSMISIÓN NEUROMUSCULAR
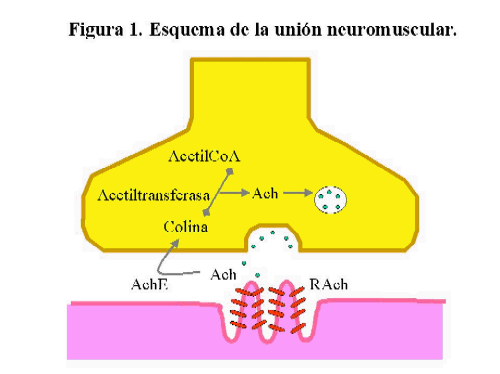
La unión neuromuscular es la zona de contacto entre la fibra nerviosa terminal y la membrana especializada de la fibra muscular (fig1). El transmisor químico es la acetilcolina (Ach), sintetizada en la terminación nerviosa a partir de acetil-CoA y colina por la colina-acetiltransferasa y almacenada en vesículas sinápticas en forma de cuantos en cantidades de 5.000 a 10.000 moléculas de Ach 6. Estas vesículas se agrupan en sitios específicos de la membrana presináptica denominados zonas activas.
En reposo, existe una liberación espontánea cuántica intermitente con una frecuencia de 1-5/seg, correspondiente a exocitosis de vesículas, que depende de la concentración de calcio extracelular y de la temperatura 7-8. Los cuantos de Ach liberada pueden unirse a la glicoproteína de la membrana postsináptica denominada receptor para la acetilcolina (RAch) abriendo durante unos pocos milisegundos las vías de iones de RAch y dando por resultado breves despolarizaciones de la membrana en la union neuromuscular que se denominan potencial miniatura de placa motora (PMPM). A los pocos segundos la Ach es hidrolizada por la acetilcolinesterasa en ácido acético y colina .
La llegada de un potencial de acción a la terminal nerviosa va a producir la apertura de los canales de calcio sensibles al voltaje con un aumento de la concentración de este ion en la terminal nerviosa dando como resultado la liberación de más de 100 cuantos de Ach que en condiciones normales permite suficiente número de uniones con el receptor para producir la aparición del potencial de placa motora (PPM). La cantidad de cuantos liberados va a depender fundamentalmente de las vesículas disponibles para liberación inmediata y de la concentración de Ca+ 9. La amplitud del PPM en condiciones normales es suficiente para superar el valor umbral y desencadenar el potencial de acción que puede ser transmitido a lo largo de la membrana muscular y dar lugar a la contracción .
El exceso de amplitud del PPM es lo que se denomina factor de seguridad de la transmisión neuromuscular que dependerá de los procesos de liberación de Ach, de la síntesis de la misma a partir de la actividad de la Ach-E y del estado funcional de los RACh.
Este factor de seguridad va a ser modificado tras estimulación nerviosa o tras ejercicio. Así, a frecuencias de estímulo elevadas o ejercicio breve va a existir un incremento del margen de seguridad por aumento de la entrada de Ca+ en el terminal con aumento de los cuantos de ACh liberada, fenómeno denominado Facilitación postactivación. Cuando los estímulos se realizan con intervalos mayores, a frecuencias entre 2-3 Hz, el factor de seguridad para el segundo estímulo va a ser menor, con disminución del PPM debido probablemente a una disminución de vesículas con disposición inmediata para la liberación y a la meseta hasta el equilibrio entre liberación y movilización cuántica.
Estas variaciones del factor de seguridad, no van a tener repercusión clínica y o electrofisiológica en uniones neuromusculares con función normal, pero sí en aquellos procesos que alteren la liberación cuántica, la síntesis de Ach o conlleven una disminución de Rach, conformando los dos grupos fisiopatológicos de transtornos, los de origen presináptico y postsináptico 5,9 y que serán la base de las alteraciones electrofisiológicas.
TÉCNICAS ELECTROFISIOLÓGICAS
Estimulación repetitiva
Esta basada en el estudio de la amplitud o área del potencial motor evocado (PME) tras estimulación nerviosa simple o repetitiva a diferentes frecuencias, como medida del número de fibras activadas por el impulso nervioso 10. Para la evaluación segura y precisa de estos cambios es fundamental el control de errores técnicos utilizando electrodos de registro de superfice, asegurando estimulaciones supramaximales, controlando variaciones de la temperatura y artefactos de movimiento.
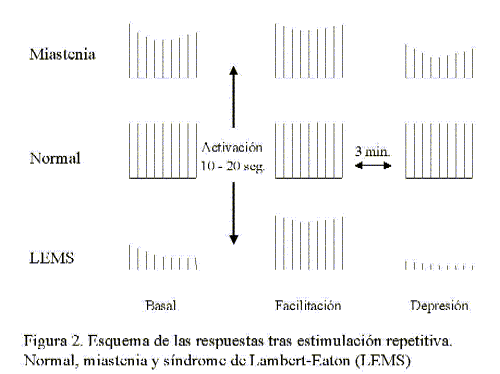
En la aplicación de bajas frecuencias, entre 2-3 Hz, se va a valorar el decremento del 4º-5º potencial respecto al primero (que coincide con el momento en que el factor de seguridad es menor) cuyo resultado en personas normales no disminuye más de 5-8% 10-11. En la mayoría de transtornos con alteración pre- o postsináptica habrá un decremento mayor que traduce el número de fibras musculares que sufren bloqueos (fig 2).
Después de un período de contracción máxima o de estimulación repetitiva a altas frecuencias la amplitud y el área del PME no va alterarse en músculos normales. En el miasténico evocará una respuesta de mayor amplitud con un decremento menor del 4º-5º potencial 10. Este efecto de facilitación de la liberación de Ach es particularmente pronunciado y característico de los transtornos presinápticos como el síndrome de Lambert-Eaton (LEMS). Un test simple que traduce el fenómeno de potenciación postetánica en estos transtornos es la valoración del PME aislado, previo y tras ejercicio breve 12.
La facilitación postetánica es transitoria y va a ser seguida entre el 2º y 4º minuto de una disminución mayor de la amplitud en el músculo miasténico denominada depresión postetánica, consecuecia de una reducción mayor del margen de seguridad. Su base fisiopatológica no está totalmente aclarada implicándose una disminución de la reserva de la Ach disponible para la liberación 12 y/o desensibilización de los receptores 10 (fig.3).
Otras estrategias que modifican el factor de seguridad, como son el aumento de la temperatura y la isquemia o la combinación de isquemia y ejercicio con técnicas de estimulación repetitiva, son utilizadas para aumentar el rendimiento diagnóstico en músculos donde los estudios convencionales pueden ser negativos 13.
Electromiografía de fibra única (SFEMG)
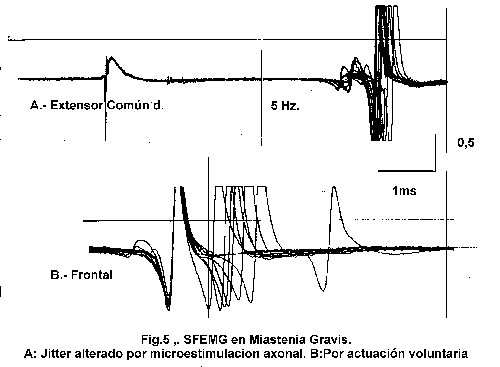
El desarrollo de la EMG de fibra simple a partir de Erïk Stälberg y Jan Ekstedt 14 ha permitido el estudio de la microfisiología de la unidad motora mostrando el espectro de los cambios funcionales en placas motoras individuales. Un electrodo especial de 25m m de diámetro con un área de registro de 3 m m 15 va a permitir el registro de los potenciales de acción de dos o más fibras musculares de una unidad motora activada voluntariamente, valorando el número de fibras por registro o densidad de fibras y la variación existente entre los intervalos de estos potenciales en las sucesivas descargas o Jitter, medido como la media de las diferencias consecutivas (MCD). Esta variación reflejará fundamentalmente el tiempo que se requiere para que los PPM en la unión neuromuscular alcancen el umbral para generar el potencial de acción, siendo una medida sensible del factor de seguridad de la transmisión neuromuscular. Cuando el PPM es insuficiente para llegar al umbral se obtendrán bloqueos del potencial de fibra muscular 14. Cuando el bloqueo ocurre en varias placas terminales de un músculo se manifestará la debilidad clinica 5.
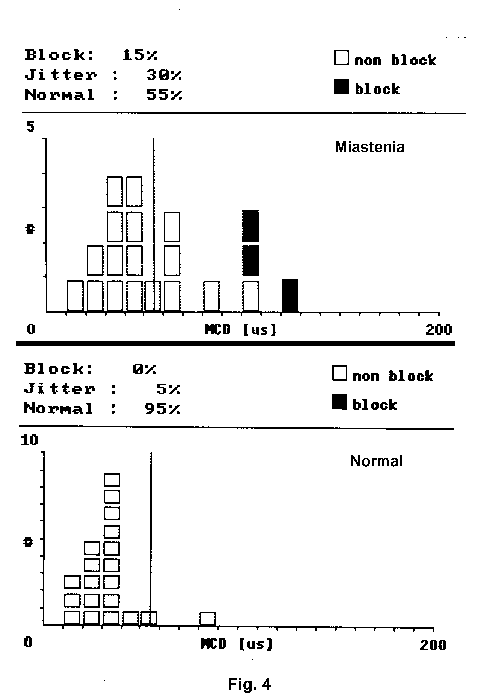
En condiciones normales los valores de fluctuación van a variar de unos músculos a otros y de unas placas a otras dentro del mismo músculo, por lo que un muestreo adecuado se basa en la valoración de 20 pares dentro de un mismo músculo (fig. 4). Estos valores de referencia, para los diferentes músculos y grupos de edad, estan reflejados en un estudio multicéntrico 16,17.
El criterio de anormalidad es la obtención de al menos un 10% de placas con aumento del Jitter, aumento del Jitter medio por encima del límite superior del músculo y la presencia de bloqueos en alguno de ellos 15,16 (fig. 4).
El estudio de Jitter puede realizarse mediante microestimulación eléctrica siendo útil en pacientes en los que la cooperación es difícil (niños de corta edad, estados comatosos o pacientes con temblor) o en aquellos estudios que deba controlarse la frecuencia de activación 15. El resultado del Jitter mediante esta técnica será menor, dada la valoración de placas individuales, siendo la relación entre los dos la siguiente:
MCD medio (estim. axonal) = MCD medio (activ. voluntaria) /
EMG convencional
Va a ser utilizado para excluir o confirmar la presencia de afectación concomitante de nervio o músculo, diferenciando el transtorno neuromuscular secundario a dicha alteración. En alteraciones primarias podemos obtener variabilidad de la morfología y amplitud de los potenciales de unidad motora con acortamiento y morfología polifásica en relación con el grado de fatiga 5,10.
RENDIMIENTO DIAGNÓSTICO DEL ESTUDIO ELECTROFISIOLÓGICO
El rendimiento diagnóstico de las diferentes técnicas y su elección en la miastenia gravis (MG) va a depender de la forma clínica de la enfermedad 18-19, lo que condicionará su posible aplicación a territorios musculares que expresen fatigabilidad. Así, los estudios de estimulación repetitiva seran más anormales cuando se exploran músculos proximales 10,20 o se realizan test de activación en los distales 21. Los hallazgos más característicos 22 van a ser:
- Disminución de la amplitud o área del 4º-5º potencial a bajas frecuencias (2-5 Hz).
- Aumento del decremento en la fase de agotamiento post-activación.
- Menos característico es el aumento de la primera respuesta con menor decremento en el período de facilitación postetánica, pues está en relación con la amplitud del potencial basal.
El estudio de SFEMG mediante activación o microestimulación axonal se ha demostrado más sensible que el resto de técnicas 2,7,15,23-24 detectando alteraciones en alto grado en función de la forma clínica y músculo explorado; siendo de elección el extensor digitorum communis en las formas generalizadas y el frontalis u orbicularis oculi en las oculares (fig5).
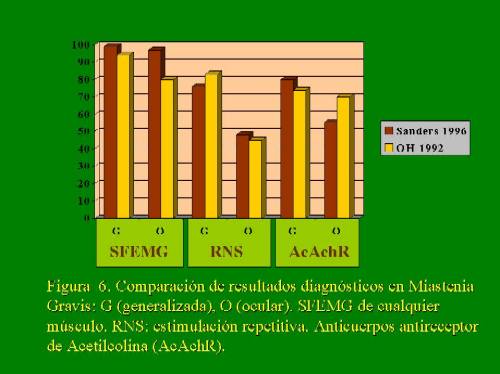
Esta mayor sensibilidad limita la especificidad, ya que se ha demostrado su alteración en otras patologías de nervio y músculo 15,25. Por lo que el estudio de EMG convencional y de conducciones debe ser realizado para su diferenciación. Por el contrario, el hallazgo normal del Jitter en un músculo débil descarta alteración de la transmisión neuromuscular como causa 26. Esto va a contrastar con la mayor especificidad de los estudios inmunológicos, que a su vez tendrán como resultado un mayor porcentaje de falsos negativos 15 (fig.6). Los resultados de todas estas técnicas pueden ayudar a la caracterización de los diferentes subtipos 27.
Por otro lado, medidas seriadas del Jitter en pacientes con miastenia pueden ser de utilidad como apoyo del cuadro clínico de progresión. Así, variaciones del Jitter medio (MCD) mayores de un 10% se correlacionan con un empeoramiento o mejoría en un 60 % y 80 % de enfermos 28 respectivamente, pudiendo expresarse también esta variación por el porcentaje de placas con bloqueos 5, pero siempre relacionándola con la evolución clínica.
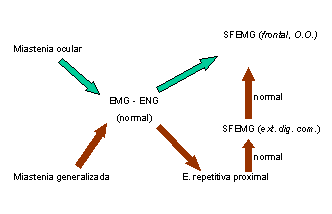
La estrategia de estudio que seguimos en nuestro laboratorio es:
Sindrome de Eaton-Lambert.
Enfermedad cuya alteración inmunológica viene marcada por la presencia de anticuerpos frente a canales de calcio, asociado frecuentemente a cáncer de pulmón y dando lugar a un transtorno presináptico de la unión neuromuscular, que se manifiesta por clínica de debilidad proximal, transtornos autonómicos y reflejos disminuidos 29-30.
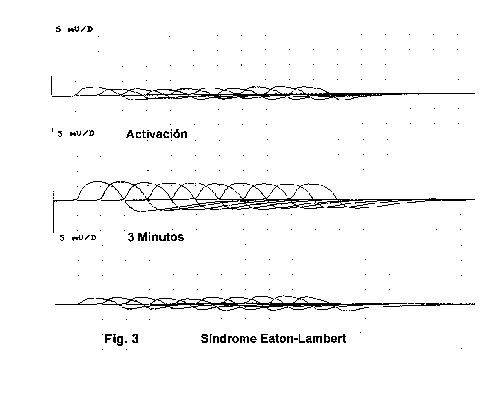
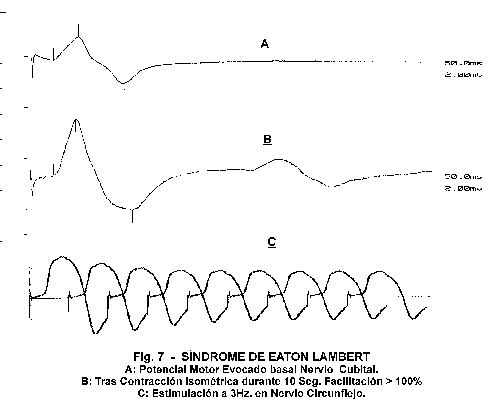
Los resultados electrrofisiológicos de estimulación repetitiva en estos enfermos (fig7) muestran un comportamiento característico 4,30-31:
- Reducción del PME obtenido tras estímulo simple.
- Aumento de la amplitud y área del PME obtenido tras contracción maximal durante 10-30" en resultados superiores al 100%.
- La estimulación a bajas frecuencias va a mostrar un patrón decremental progresivo con facilitación o incremento a altas frecuencias.
Estas alteraciones van a distribuirse en grado variable en los diferentes músculos, siendo en general más sensibles los distales 4 y el anconeo 32, aunque en ocasiones puede ser necesario la elección de músculos específicos33-34 más sintomáticos
El Jitter va a estar mas alterado que en la miastenia 15, llegando a porcentajes del 100% en los enfermos de algunas series 30. Los bloqueos mejoran cuando aumenta la frecuencia de activación o de estimulación de 2 a 10-20 Hz 15; fenómeno objetivado también en algunas de las placas de músculos miasténicos 35.
La EMG convencional va a mostrar una alteración de la estabilidad de los potenciales de unidad motora (PUM) marcada y neuropatías asociadas al síndrome paraneoplásico 30.
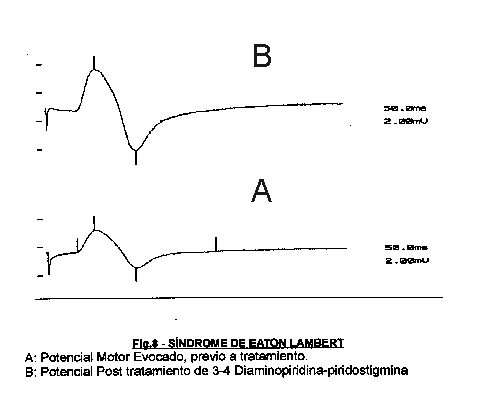
El método de elección en el diagnóstico de estos procesos es la valoración del potencial motor evocado previa e inmediatamente después de la tetanización por contracción maximal durante 10 sg 4,36 en varios músculos por su alto rendimiento diagnóstico y las menores molestias para el enfermo en relación con la estimulación a altas frecuencias. Por otro lado, el comportamiento del PME va a ser de utilidad también en el seguimiento y valoración terapéutica de estos enfermos 37-38 (fig 8).
No existe en todos los casos una clara diferenciación electrofisiológica y clínica entre MG y LEMS, objetivando en músculos miasténicos hallazgos característicos de LEMS 39 y en algunos LEMS hallazgos de MG 31 como expresión fisiopatológica de diferentes subtipos. Por otro lado, esta descrita la coexistencia clínica e inmunológica de estos síndromes 40-41.
Síndromes miasténicos congénitos
Los síndromes miasténicos congénitos son transtornos poco frecuentes, sin anormalidades inmunológicas, que pueden manifestarse de forma severa desde el nacimiento o con escasa y variable expresión clínica. Los estudios morfológicos y electrofisiológicos in vitro y los estudios genéticos de los síndromes postsinápticos son los que han ayudado en los ultimos años a su caracterización 42-43.
Los estudios electrofisiológicos en estos síndromes van a mostrar de forma global una respuesta decreciente a la estimulación repetitiva a bajas frecuencias 42 en músculos débiles. La existencia de fluctuaciones episódicas con intervalos clínicos de normalidad, característico de la miastenia familiar infantil puede normalizar las respuestas, activando dicha alteración con estimulación nerviosa a frecuencias entre 3-5 Hz durante 5 minutos, que condicionará una respuesta decreciente en los siguientes minutos 44.
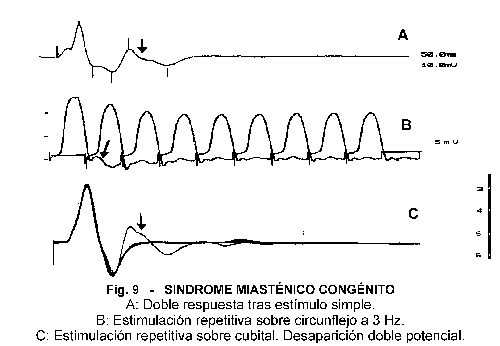
Los estímulos aislados van a mostrar potenciales de amplitud normal con dobles respuestas en los síndromes clásicos de canal lento y de la deficiencia de ACh-E 45-46. Estas descargas van a desaparecer durante la estimulación repetitiva o tras la contracción maximal, por lo que deben obtenerse con el músculo en reposo y fuera de los efectos de los inhibidores de la AChE (Fig 9).
La electomiografía convencional va a mostrar patrón miopático 45,47 con potenciales de unidad motora inestables. La SFEMG va a confirmar la presencia de un transtorno de transmisión neuromuscular sin resultados diferenciales 48.
Otros transtornos de transmisión neuromuscular
Diferentes toxinas, farmacos, picaduras o venenos de animales van a alterar la transmisión neuromuscular en nivel pre- o postsináptico, dando lugar a un comportamiento electrofisiológico acorde con la fisiopatologia 49.
Cabe destacar entre los síndromes presinápticos el botulismo con expresión electrofisiológica similar al síndrome de Eaton-Lambert en el que existirá una disminución de amplitud del PME tras estímulo simple en músculos afectos con un menor grado de facilitación postetánica sin depresion postactivación, siendo infrecuente el hallazgo decremental a bajas frecuencias50-51.
La intoxicación por organofosforados va a ser otra de las causas graves de alteración de la tansmisión neuromuscular debida a una inhibición de la colinesterasa, que se traducirá por un decremento a frecuencias rápidas y actividad repetitiva tras estímulo simple como en algunos síndromes miasténicos congénitos 52.
Bibliografía
- Howard J.F,Sanders D B, Massey J.M:Electrodiagnostico de miastenia gravis y sindrome miasténico de Lambert -Eaton.Clinicas Neurologicas de Norteamérica 1994 2:283-307
- Sanders D B,Stälberg E V. AAEM Minimonograph # 25: Single-Fiber electromyography. Muscle Nerve. 1996; 19:1069-1083.
- Jablecki Ch K. AAEM Case report # 3: Myasthenia gravis. Muscle Nerve.1991;14:391-397.
- Maddison P.,Newsom davis J. Distribution of electrophysiological abnormality in Lambert Eaton myastenic syndrome. J. Neurol. Neurosurg. Psychriaty. 1998;65:213-217.
- Cruz Martínez A., Montero J. EMG fisiopatología de la transmisión neuromuscular. En Diez Tejedor E (ed.). Miastenia gravis y síndromes miasténicos. J. R. Prous. Barcelona 1995; 17-75.
- Brown W F. Neuromuscular transmission normal and abnormal. In Butterworth (eds).The physiological and technical basis of electromyography. 1ªed. USA; 1984 p.369-427
- Keesey J C.AAEE Minimonograph # 33: Electrodiagnostic approch to defects of neuromuscular transmision. Muscle Nerve 1989; 12 613-626
- Magleby K L. Neuromuscular transmision. In Engel A G. Banker B Q (eds): Myology Basic and Clinical, New York. Macgraw Hill 1986 Vol 1 p: 393-418
- Maselli R A. Fisiopatologia de la miastenia gravis y síndrome de Lambert-Eaton. Clínicas Neurológicas de Norteamérica 1994; 2: 265-282.
- Stalberg E. Clinical electrophysiology in myastenia gravis. J. Neurol. Neurosurg. Psychiatry 1980; 43: 622-633.
- Desmedt, J E. The neuromuscular disorder in myasthenia gravis:Electrical and mechanical responses to nerve stimulation in hand muscles. In Desmedt, J.E. (de). New Developements in electromyography and clinical neurophysiology. Karger, Basel 1973;1:241-304.
- Kimura J.Tecniques of repetitive stimulation. In F.A. Davis Philadelphia 1983 p:189-203
- Gilchrist J.M. "Double-step" repetitive stimulation in myasthenia gravis. Muscle Nerve 1987; 10:233-237.
- Stälberg, E Ekstedt, J.Single fibre electromyography and mycrophysiology of the motor unit in normla and diseased muscle. En: Desmedt, J.E. (ed). New developments in electromyography and clinical neurphysiology. Karger, Basel 1973, 1:113-179.
- Stalberg E Trontelj JV. Single Fiber Electromyography. Studies in Healthy and diseased muscle. 2nd ed. Raven Press, New York 1994.
- Gilchrist J.M.:Single fiber EMG reference values: a collaborative effort. Report from the ad hoc commitee of the AAEM special interest group on single fiber EMG. Muscle Nerve 1992; 15: 151-161
- Bromberg, M. B., Scott D.M. Single fiber EMG: reference values reformatted in tabular form. Muscle Nerve 1994; 17: 820-821.
- Sanders DB, Howard JF, AAEE Minimonograff # 25: Single fiber electromyogrphy in myasthenia gravis. Muscle nerve 1986 ; 9: 809-819.
- Maher J, Gran'maison F, Nicolle MW, Strong MJ, Bolton ChF. Diagnostic difficulties in myasthenia gravis. Muscle Nerve 1998; 21: 577-583.
- Kimura J. Myasthenia gravis and other disorders of neuromuscular transmission. In Kimura J: Electrodiagnosis in diseases of nerve and muscle: principles and practice. F.A. Davis, Philadelphia, 1983. P:511-525
- Desmedt JE, Borenstein S. Double step nerve stimulation test for myasthenic block sensitization of post-activation exhaustion by ischemia. Ann Neurol 1977; 1: 55-64.
- Oh SJ, Eslami N, Nishihira T, et all. Electrophysiological and clinical correlation in myasthenia gravis. Ann Neurol 1982; 12: 348-354.
- Cruz Martinez A, Ferrer MT, Pérez Conde MC.y cols. Miastenia gravis. Actualización del diagnóstico electrofisiológico (II). Electromiografia de fibra única (SFEMG). Correlación con los resultados de otras técnicas diagnósticas. Rev Neurol 1981; 42: 197-211.
- Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Dignostic sensitivity of the laboratory tests in myasthenia Gravis. Muscle nerve 1992; 15: 720-724.
- Bertoni TE, Stalberg E, Yuson CP, Engel K. Single-fiber electromyogrphy in neuromuscular disorders: Correlation of muscle histochemystry, Single-fiber Electomyogrphy, and clinical findings. Muscle Nerve 1994; 17: 345-353.
- Sanders, DB, Howard JF.AAEE minimonograph # 25: Single fiber electromyography in myasthenia gravis. Muscle Nerve 1986;9: 809-819.
- Sommer N, Sigg B, Mels A; et all: Ocular Myasthenia gravis:response to long term immunosuppressive treatment. J Neurol Neurosrg Psychyatry 1997; 62:156-162.
- Sanders B. Single-fiber electromyography. Introduction to fiber density & jitter. International course on single fiber emg. September, 1998, Vigo-Spain.
- Lang B, Newson-Davis J,Wray D, Vincent A. Autoimmune aethiology for myasthenic (Eaton -Lambert) syndrome. Lancet 1981; 2:224-226.
- O'Neill JH, Murray NMF, Newson-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain 1988; 111: 577-596.
- Oh SJ, Kim DE, Kuruoglu R,Brooks J, Claussen G. Electrophysiological and clinical correlations in the Lambert-Eaton myasthenic syndrome. Muscle Nerve 1996; 19: 903-906.
- Kennet RP, Fawcet PRW. Repetitive nerve stimulation of anconeus in the assesment of neuromuscular transmisión disorders. Electroencephalogr Clin Neurophysiol 1993; 89: 170-176.
- Nicolle MW, Stewart DJ, Remtulla H,Chen R, Bolton CF. Lambert-Eaton myasthenic syndrome presenting with severe respiratory failure. Muscle Nerve 1996; 19:1328-33.
- Oh SJ, Head T, Fesenmeier J. Clausen G. Peroneal nerve repetitive nerve stimulation test: its value in diagnosis of myasthenia gravis and Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1995; 18: 867-873.
- Trontelj JV. Stälberg E. Single motor end-plates in myastenia gravis and lems at different firing rates. Muscle Nerve 1990; 14:226-232.
- Tim RW, Sanders DB. Repetitive nerve stimulation studies in the Lambert-Eaton myasthenic syndrome. Muscle Nerve 1994; 17:995-1001.
- Lundh H, Nilsson O, Rosen Y, Johansson S. Practical aspects of 3,4-diaminopyridine treatment of the labert-eaton myasthenic syndrome. Acta Neurol Scand. 1993;88: 136-140.
- Morata C, Alfaro A, Chumillas MJ, Montalar J. Síndrome miasténico de Lambert-Eaton: Respuesta al tratamiento con 3,4-diaminopiridina. Med. Clin (Bar) 1995; 104:156.
- Singer P, Smith L, Ziegler DK, Festtoff BW. Postetanic potentiation in a patient with myasthenia gravis. Neurology 1981; 31: 1345-1347.
- Newsom-Davis J. Leys K, Vicent A, Fegurson Y, Modi G,Mills K. Immunological evidence for the co-existence of the Lambert-Eaton myasthenic syndrome and myasthenia gravis in two patients. J Neurol Neurosurg Psychriatry 1991; 54: 452-453.
- Tabbaa MA, Leshner RT, Campbell W. Malignant thymoma with dysautonomia and disordered neuromuscular transmission. Arch Neurol 1986; 43:955-957.
- Engel AG. Congenital myasthenic syndromes. In Lisak RP edit. Handbook of Myasthenia gravis and myasthenic syndromes. New York; 1994 p.33-62.
- Gomez CM, Maselli R, Gammack J,Lasalde J, Tamamizu S, CornblathDR et al. A beta-subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann Neurol 1996; 39 : 712-23.
- Mora M, Lambert EH, Engel AG. Synaptic vesicle abnormality in familial infantile myasthenia. Neurology 1987; 37:206-214.
- Oosterhuis HJ,Newsom-Davis J, Wokke JH, Molenaar PC, Weerden TV, Oen BS, et all. The Slow channel syndrome. Two new cases. Brain 1987; 110: 1061-79.
- Hutchinson DO, Timothy J, Nakano S, Camp S, Taylor P, Harper M et al. Congenital endplate acetylcholinesterase deficiency. Brain 1993; 116:633-653.
- Furui E Fukushima K, Sakashita T, Sakato S,Matsubara S, Takamori M. Familial limb girdle myasthenia with tubular aggregates. Muscle Nerve 1997; 20: 2599-603.
- Engel AG ,Nagel A, Walls IV. Harper CM, Waisburg HA. Congenytal myasthenic syndromes:I Deficiencie and short open time of the acetylcholine receptor. Muscle Nerve 1993: 16:1284-92.
- Rivner MH. Swift T. Electrical testing in disorders of neuromuscular transmission. In Brown and Bolton edits.Clinical Electromyography 2nd ed.1993, Butterwort p:625-651.
- Cherington M. Clinical spectrum of botulism. Muscle Nerve 1998;21:701-710.
- Maselli R. Ellis W Mandler R. Sheikh F. Senton G. Knox S. Cluster of wound botulism in california: clinical, electrohysiologic, and pathologic study.Muscle Nerve 1997; 20:1284-1295.
- Wadia S.Chitra S. Bamin R. Kiwalkar RS. Sardesai HV. Electrophysiological studies in acute organophosphate poisoning. J. Neurol Neurosurg Psychiatr 1987; 50:1442-1448.